
POMALIDOMIDE REIG JOFRE 1 mg, gélule, boîte de 21
Dernière révision : 26/02/2025
Taux de TVA : 2.1%
Laboratoire exploitant : FORTE PHARMA
Source :

POMALIDOMIDE REIG JOFRE, en association avec le bortézomib et la dexaméthasone, est indiqué dans le traitement du myélome multiple chez les patients adultes ayant déjà reçu au moins un traitement antérieur comportant le lénalidomide.
POMALIDOMIDE REIG JOFRE, en association avec la dexaméthasone, est indiqué dans le traitement du myélome multiple en rechute et réfractaire chez les patients adultes ayant déjà reçu au moins deux traitements antérieurs comportant le lénalidomide et le bortézomib, et dont la maladie a progressé au cours du dernier traitement.
- Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
- Grossesse.
- Femmes en capacité de procréer, sauf si toutes les conditions du programme de prévention de la grossesse sont réunies (voir rubriques Mises en garde spéciales et précautions d'emploi et Fertilité, grossesse et allaitement).
- Hommes incapables de suivre ou de respecter les mesures contraceptives requises (voir rubrique Mises en garde spéciales et précautions d'emploi).
Tératogénicité
Le pomalidomide ne doit pas être pris pendant la grossesse car un effet tératogène est attendu. Le pomalidomide est structurellement proche du thalidomide. Le thalidomide est un tératogène humain connu qui provoque des anomalies congénitales sévères, pouvant engager le pronostic vital. Chez le rat et le lapin, le pomalidomide s'est révélé tératogène lorsqu'il a été administré pendant la phase d'organogenèse majeure (voir rubrique Données de sécurité préclinique).
Les conditions du programme de prévention de la grossesse doivent être remplies par toutes les patientes, à moins de pouvoir certifier que la patiente est dans l'incapacité de procréer.
Critères définissant, pour une femme, l'incapacité de procréer
Toute patiente ou partenaire d'un patient est considérée comme étant dans l'incapacité de procréer si elle présente au moins l'un des critères suivants :
- Age ≥ 50 ans et aménorrhée naturelle depuis ≥ 1 an (l'aménorrhée faisant suite au traitement d'un cancer ou pendant l'allaitement ne suffit pas à exclure un risque de grossesse)
- Ménopause précoce confirmée par un gynécologue
- Salpingo-ovariectomie bilatérale ou hystérectomie
- Génotype XY, syndrome de Turner, agénésie utérine.
Information des patients
Chez la femme en capacité de procréer, le pomalidomide est contre indiqué à moins que toutes les conditions suivantes soient réunies :
- Elle comprend les risques tératogènes attendus pour l'enfant à naître.
- Elle comprend la nécessité d'une contraception efficace, sans interruption, débutée au moins 4 semaines avant le traitement, poursuivie pendant toute la durée du traitement et au moins 4 semaines après la fin de celui-ci.
- Même en cas d'aménorrhée, toute femme en capacité de procréer doit suivre tous les conseils pour une contraception efficace.
- Elle doit être en mesure de respecter les mesures de contraception efficaces.
- Elle est informée et comprend les conséquences potentielles d'une grossesse et la nécessité de consulter rapidement un médecin s'il existe un risque de grossesse.
- Elle comprend la nécessité de commencer le traitement dès que le pomalidomide lui a été délivré suite à un test de grossesse négatif.
- Elle comprend la nécessité et accepte d'effectuer un test de grossesse au moins toutes les 4 semaines pendant le traitement, sauf en cas de stérilisation tubaire confirmée.
- Elle confirme avoir bien compris les risques et les mesures de précaution nécessaires pour l'utilisation du pomalidomide.
Pour les femmes en capacité de procréer, le médecin prescripteur doit s'assurer que :
- La patiente remplit les conditions requises par le programme de prévention de la grossesse, y compris une compréhension du risque satisfaisante.
- La patiente confirme avoir compris les conditions susmentionnées.
Pour les patients de sexe masculin traités par pomalidomide, les données pharmacocinétiques ont démontré que le pomalidomide était présent dans le sperme humain pendant le traitement. A titre de précaution, et en tenant compte de l'allongement possible du temps d'élimination dans les populations particulières telles que les patients présentant une insuffisance hépatique, tout patient de sexe masculin sous pomalidomide doit remplir les conditions suivantes :
- Comprendre les risques tératogènes attendus en cas de rapport sexuel avec une femme enceinte ou en capacité de procréer.
- Comprendre qu'il est nécessaire d'utiliser des préservatifs en cas de rapport sexuel avec une femme enceinte ou en capacité de procréer n'utilisant pas une méthode de contraception efficace pendant toute la durée du traitement, en cas d'interruption des prises et pendant 7 jours après l'interruption et/ou l'arrêt du traitement. Cela concerne également les hommes vasectomisés qui doivent utiliser un préservatif en cas de rapport sexuel avec une femme enceinte ou en capacité de procréer car le pomalidomide peut être présent dans le liquide séminal malgré l'absence de spermatozoïdes.
- Comprendre qu'en cas de survenue d'une grossesse chez sa partenaire pendant le traitement par pomalidomide ou pendant 7 jours après l'arrêt du traitement, il doit informer immédiatement son médecin traitant qui devra recommander d'adresser sa partenaire à un médecin spécialiste ou expérimenté en tératologie pour évaluation et conseil.
Contraception
Les femmes en capacité de procréer doivent utiliser au moins une méthode de contraception efficace pendant au moins 4 semaines avant le début du traitement, pendant toute la durée de celui-ci et au moins 4 semaines après l'arrêt du traitement par pomalidomide, même en cas d'interruption du traitement, à moins qu'elles ne déclarent une abstinence totale et continue, qui sera confirmée de façon mensuelle. Si la patiente n'utilise aucune méthode contraceptive efficace, elle devra être orientée vers un médecin compétent pour être conseillée et afin qu'une contraception soit instaurée.
Voici des exemples de méthodes de contraception adaptées :
- Implant contraceptif
- Dispositif intra-utérin au lévonorgestrel
- Acétate de médroxyprogestérone retard
- Stérilisation tubaire
- Rapports sexuels exclusivement avec un partenaire vasectomisé ; la vasectomie doit avoir été confirmée par deux spermogrammes négatifs
- Pilule progestative inhibant l'ovulation (c'est-à-dire, désogestrel)
En raison du risque accru de thromboembolie veineuse chez les patients atteints de myélome multiple et traités par l'association pomalidomide et dexaméthasone, l'utilisation de pilules œstro-progestatives n'est pas recommandée (voir également rubrique Interactions avec d'autres médicaments et autres formes d'interactions). Si la patiente est sous pilule œstro-progestative, elle devra changer de méthode contraceptive, c'est à dire utiliser l'une des méthodes susmentionnées. Le risque dethromboembolie veineuse persiste pendant 4 à 6 semaines après l'arrêt du contraceptif oral œstro-progestatif. L'efficacité des contraceptifs stéroïdiens peut être diminuée en cas de traitement concomitant par dexaméthasone (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Les implants contraceptifs et les dispositifs intra-utérins au lévonorgestrel sont associés à un risque accru d'infection lors de leur insertion et à des saignements vaginaux irréguliers. Le recours aux antibiotiques à titre prophylactique doit être envisagé, en particulier chez les patientes présentant une neutropénie.
Les dispositifs intra-utérins au cuivre ne sont pas recommandés en raison des risques potentiels d'infection lors de leur insertion et des règles abondantes qu'ils peuvent entraîner, susceptibles de mettre en danger les patientes présentant une neutropénie sévère ou une thrombopénie sévère.
Tests de grossesse
Selon les pratiques locales, et sous la responsabilité d'un médecin, les femmes en capacité de procréer doivent se soumettre à des tests de grossesse d'une sensibilité minimale de 25 mUI/mL, comme indiqué ci-dessous. Cette obligation s'applique également aux femmes en capacité de procréer pratiquant une abstinence totale et continue. Idéalement, le test de grossesse, la prescription et la délivrance du médicament auront lieu le même jour. La délivrance du pomalidomide aux femmes en capacité de procréer doit avoir lieu dans les 7 jours suivant la prescription.
Avant de commencer le traitement
Un test de grossesse doit être effectué sous contrôle médical lors de la consultation ou dans les 3 jours précédant la consultation au cours de laquelle le pomalidomide est prescrit si la patiente utilise une méthode de contraception efficace depuis au moins 4 semaines. Le test doit confirmer que la patiente n'est pas enceinte au moment où elle débute le traitement par pomalidomide.
Suivi et arrêt du traitement
Un nouveau test de grossesse sous contrôle médical doit être effectué au moins toutes les 4 semaines et au moins 4 semaines après l'arrêt du traitement, sauf en cas de stérilisation tubaire confirmée. Ces tests de grossesse doivent être effectués le jour de la consultation dédiée à la prescription ou dans les 3 jours précédents.
Précautions supplémentaires
Les patients doivent être informés de ne jamais donner ce médicament à quelqu'un d'autre et de rapporter les gélules non utilisées à leur pharmacien en fin de traitement.
Les patients ne doivent pas faire de don de sang ou de sperme pendant le traitement (y compris pendant les interruptions de traitement) et pendant 7 jours après l'arrêt du traitement par pomalidomide.
Les professionnels de santé et les aidants doivent porter des gants à usage unique pour manipuler la plaquette ou la gélule. Les femmes enceintes ou qui pensent l'être ne doivent pas manipuler la plaquette ou la gélule (voir rubrique Précautions particulières d’élimination et de manipulation).
Guide d'aide à la prescription, restrictions de prescription et de délivrance
Afin d'aider les patients à éviter toute exposition du fœtus au pomalidomide, le titulaire de l'autorisation de mise sur le marché (AMM) fournira aux professionnels de santé des documents explicatifs visant à renforcer les mises en garde relatives à la tératogénicité attendue du pomalidomide, à donner des conseils concernant la mise en place d'une contraception préalable au traitement et à fournir des explications sur les tests de grossesse nécessaires. Le prescripteur doit informer le patient du risque tératogène attendu et des mesures contraceptives strictes définies dans le programme de prévention de la grossesse et lui remettre la brochure appropriée d'information pour les patients, la carte patient et/ou un document équivalent, tel que défini avec chaque autorité compétente nationale. En collaboration avec chaque autorité compétente nationale, un programme d'accès contrôlé a été mis en place ; il inclut l'utilisation d'une carte patient et/ou d'un document équivalent pour le contrôle des prescriptions et/ou des délivrances et le recueil d'information relatives à l'indication afin de surveiller l'utilisation hors AMM sur le territoire national. Dans l'idéal, le test de grossesse, la prescription et la délivrance du médicament doivent avoir lieu le même jour. La délivrance du pomalidomide aux femmes en capacité de procréer doit avoir lieu dans les 7 jours suivant la prescription et après un test de grossesse négatif effectué sous contrôle médical. La prescription doit être limitée à une durée de traitement de 4 semaines maximum, conformément aux schémas posologiques dans les indications autorisées (voir rubrique Posologie et mode d'administration) chez les femmes en capacité de procréer et de 12 semaines maximum chez tous les autres patients.
Evénements hématologiques
L'effet indésirable hématologique de grade 3 ou 4 le plus fréquemment rapporté chez les patients présentant un myélome multiple en rechute/réfractaire a été la neutropénie, suivie de l'anémie et de la thrombopénie. Les patients doivent faire l'objet d'une surveillance en vue de détecter l'apparition d'effets indésirables hématologiques, en particulier d'une neutropénie, et doivent être informés de la nécessité de signaler rapidement tout épisode fébrile. Les médecins doivent surveiller les patients en vue de détecter des signes évocateurs d'une hémorragie, y compris les épistaxis, notamment en cas d'utilisation de médicaments concomitants connus pour majorer le risque de saignements (voir rubrique Effets indésirables). L'hémogramme complet doit être contrôlé à l'inclusion, une fois par semaine pendant les 8 premières semaines, puis une fois par mois. Une modification de dose peut être nécessaire (voir rubrique Posologie et mode d'administration). Des transfusions et/ou l'administration de facteurs de croissance peuvent être nécessaires.
Evénements thromboemboliques
Des patients recevant le pomalidomide, soit en association avec le bortézomib et la dexaméthasone, soit en association avec la dexaméthasone, ont développé des événements thromboemboliques veineux (essentiellement thrombose veineuse profonde et embolie pulmonaire) et des événements thrombotiques artériels (infarctus du myocarde et accident vasculaire cérébral) (voir rubrique Effets indésirables). Les patients présentant des facteurs de risque connus de thromboembolie, notamment des antécédents de thrombose, doivent faire l'objet d'une surveillance étroite. Des mesures doivent être prises pour essayer de réduire au minimum tous les facteurs de risque modifiables (par exemple, le tabagisme, l'hypertension et l'hyperlipidémie). Il est conseillé aux patients et à leurs médecins d'être attentifs aux signes et symptômes de thromboembolie. Les patients doivent être informés qu'ils doivent consulter un médecin en cas de survenue de symptômes tels qu'un essoufflement, une douleur thoracique, un gonflement des bras ou jambes. Sauf s'il est contre indiqué, un traitement anticoagulant (par exemple, acide acétylsalicylique, warfarine, héparine ou clopidogrel) est recommandé, en particulier chez les patients présentant d'autres facteurs de risque de thrombose. La décision de mettre en place des mesures prophylactiques antithrombotiques doit être prise après une évaluation attentive des facteurs de risque sous-jacents propres à chaque patient. Au cours des études cliniques, les patients ont reçu de l'acide acétylsalicylique à titre prophylactique ou un autre traitement anti thrombotique. L'utilisation d'agents érythropoïétiques entraîne un risque d'événements thrombotiques, y compris de thromboembolie. Par conséquent, les agents érythropoïétiques et les autres médicaments susceptibles d'augmenter le risque d'événements thromboemboliques doivent être utilisés avec prudence.
Affections thyroïdiennes
Des cas d'hypothyroïdie ont été rapportés. Un contrôle optimal des comorbidités influençant la fonction thyroïdienne est recommandé avant l'instauration du traitement. Un contrôle de la fonction thyroïdienne est recommandé à l'inclusion et régulièrement par la suite.
Neuropathie périphérique
Les patients présentant une neuropathie périphérique de grade ≥ 2 n'ont pas été inclus dans les études cliniques portant sur le pomalidomide. Les précautions appropriées doivent être prises lorsque le traitement par pomalidomide est envisagé chez ces patients.
Dysfonctionnement cardiaque sévère
Les patients présentant un dysfonctionnement cardiaque sévère (insuffisance cardiaque congestive [classe III ou IV de la New York Heart Association], infarctus du myocarde au cours des 12 mois précédant le début de l'étude, angor instable ou mal contrôlé) n'ont pas été inclus dans les études cliniques portant sur le pomalidomide. Des événements cardiaques, y compris des cas d'insuffisance cardiaque congestive, d'œdème pulmonaire et de fibrillation auriculaire (voir rubrique Effets indésirables) ont été rapportés, principalement chez des patients présentant une cardiopathie préexistante ou des facteurs de risque cardiaque. Les précautions appropriées incluant la surveillance régulière de la survenue de signes ou symptômes évocateurs d'un événement cardiaque doivent être prises lorsque le traitement par pomalidomide est envisagé chez ces patients.
Syndrome de lyse tumorale
Les patients présentant le plus grand risque de syndrome de lyse tumorale sont ceux ayant une charge tumorale élevée avant le traitement. Ces patients doivent faire l'objet d'une surveillance étroite et les précautions appropriées doivent être prises.
Cancers secondaires au traitement
Des cancers secondaires, par exemple des cancers cutanés non mélanocytaires, ont été rapportés chez des patients recevant le pomalidomide (voir rubrique Effets indésirables). Les médecins doivent évaluer soigneusement les patients avant et pendant le traitement, à l'aide des méthodes conventionnelles de dépistage des cancers, pour surveiller le développement de cancers secondaires et instaurer un traitement le cas échéant.
Réactions allergiques et réactions cutanées sévères
Des cas d'angiœdème, de réaction anaphylactique et de réactions cutanées sévères, telles que le SSJ, la NET et le syndrome DRESS ont été rapportés en cas d'utilisation du pomalidomide (voir rubrique Effets indésirables). Les professionnels de santé doivent informer les patients des signes et symptômes de ces réactions et leur recommander de consulter immédiatement un médecin dès l'apparition de ces symptômes. Le traitement par pomalidomide doit être arrêté en cas de rash bulleux ou avec exfoliation ou de suspicion de SSJ, NET ou syndrome DRESS, et ne doit pas être repris après la résolution de ces réactions. Les patients présentant des antécédents de réactions allergiques graves associées au thalidomide ou au lénalidomide n'ont pas été inclus dans les études cliniques. Ces patients peuvent présenter un risque accru de réactions d'hypersensibilité et ne doivent pas recevoir le pomalidomide. L'interruption ou l'arrêt du traitement par pomalidomide doit être envisagé(e) en cas de rash cutané de grade 2-3. Le traitement par pomalidomide doit être arrêté de façon permanente en cas d'angiœdème et de réaction anaphylactique.
Sensations vertigineuses et confusion
Des sensations vertigineuses et un état confusionnel ont été rapportés avec le pomalidomide. Les patients doivent éviter les situations dans lesquelles les sensations vertigineuses ou la confusion peuvent constituer un problème et ne doivent pas prendre d'autres médicaments susceptibles de provoquer des sensations vertigineuses ou une confusion sans avis médical préalable.
Pneumopathie interstitielle diffuse (PID)
Des cas de PID et des événements associés, tels que des cas de pneumonie, ont été observés avec le pomalidomide. En cas d'apparition subite ou d'aggravation inexpliquée de symptômes pulmonaires, une évaluation attentive du patient doit être réalisée afin d'exclure un diagnostic de PID. Le traitement par pomalidomide doit être interrompu pendant l'évaluation de ces symptômes et si une PID est diagnostiquée, un traitement approprié doit être instauré. Le traitement par pomalidomide ne doit être repris qu'après une évaluation attentive du rapport bénéfice/risque.
Affections hépatiques
Des élévations importantes des taux d'alanine aminotransférase et de bilirubine ont été observées chez des patients traités par pomalidomide (voir rubrique Effets indésirables). Des cas d'hépatite nécessitant l'arrêt du traitement par pomalidomide ont également été rapportés. Il est recommandé de contrôler régulièrement les paramètres de la fonction hépatique pendant les 6 premiers mois de traitement par pomalidomide, puis en fonction des données cliniques du patient par la suite.
Infections
De rares cas de réactivation de l'hépatite B ont été rapportés à la suite du traitement par pomalidomide en association avec la dexaméthasone chez des patients présentant des antécédents d'infection par le virus de l'hépatite B (VHB). Certains de ces cas ont évolué vers une insuffisance hépatique aiguë et ont entraîné l'arrêt du traitement par pomalidomide. La sérologie VHB doit être déterminée avant l'instauration du traitement par pomalidomide. Chez les patients ayant un résultat positif au dépistage du virus de l'hépatite B, une consultation chez un médecin spécialisé dans le traitement de l'hépatite B est recommandée. La prudence est de mise en cas d'administration de pomalidomide en association avec la dexaméthasone chez des patients préalablement infectés par le VHB, y compris chez les patients présentant une sérologie positive pour les anticorps anti-HBc mais négative pour l'AgHBs. Ces patients doivent faire l'objet d'une surveillance étroite tout au long du traitement afin de détecter les signes et symptômes d'une infection active par le VHB.
Leucoencéphalopathie multifocale progressive (LEMP)
Des cas de leucoencéphalopathie multifocale progressive, dont certains d'issue fatale, ont été rapportés avec le pomalidomide. Ces cas de LEMP ont été rapportés de plusieurs mois à plusieurs années après le début du traitement par pomalidomide. Ces cas ont été généralement rapportés chez les patients prenant, de façon concomitante, de la dexaméthasone ou à la suite d'un traitement antérieur par d'autres chimiothérapies immunosuppressives. Les médecins doivent surveiller les patients à intervalles réguliers et un diagnostic différentiel de LEMP doit être envisagé chez les patients présentant de nouveaux signes ou symptômes cognitifs ou comportementaux ou des symptômes neurologiques ou une aggravation de ces signes ou symptômes. Il convient également de conseiller aux patients d'informer leur conjoint ou leurs aidants de leur traitement, ceux-ci pouvant remarquer des symptômes dont les patients ne sont pas conscients.
Le diagnostic d'une LEMP doit reposer sur un examen neurologique, une imagerie par résonance magnétique du cerveau et un dosage de l'ADN du virus JC (JCV) dans le liquide céphalo-rachidien par réaction en chaîne par polymérisation (PCR) ou une biopsie cérébrale suivie d'un test de dépistage du JCV. Une analyse négative du JVC par PCR ne permet pas d'écarter une LEMP. Une surveillance et des analyses complémentaires seront éventuellement justifiées si un diagnostic alternatif ne peut être établi.
Si une LEMP est suspectée, le traitement doit être suspendu jusqu'à ce que la LEMP soit exclue. Si la LEMP est confirmée, le pomalidomide doit être arrêté de façon permanente.
Teneur en sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par gélule, c'est-à-dire qu'il est essentiellement « sans sodium ».
Teneur en isomalt
Les patients présentant des problèmes héréditaires rares d'intolérance au fructose ne doivent pas prendre ce médicament.
Résumé du profil de sécurité
Le pomalidomide en association avec le bortézomib et la dexaméthasone
Les affections hématologiques et du système lymphatique les plus fréquemment rapportées ont été la neutropénie (54,0 %), la thrombopénie (39,9 %) et l'anémie (32,0 %). Les autres effets indésirables les plus fréquemment rapportés ont inclus neuropathie périphérique sensitive (48,2 %), fatigue (38,8 %), diarrhée (38,1 %), constipation (38,1 %) et œdème périphérique (36,3 %). Les effets indésirables de grade 3 ou 4 les plus fréquemment rapportés ont été des affections hématologiques et du système lymphatique incluant neutropénie (47,1 %), thrombopénie (28,1 %) et anémie (15,1 %). L'effet indésirable grave le plus fréquemment rapporté a été la pneumonie (12,2 %). Les autres effets indésirables graves rapportés ont été la fièvre (4,3 %), l'infection des voies aériennes inférieures (3,6 %), la grippe (3,6 %), l'embolie pulmonaire (3,2 %), la fibrillation auriculaire (3,2 %) et l'insuffisance rénale aiguë (2,9 %).
Le pomalidomide en association avec la dexaméthasone
Les effets indésirables les plus fréquemment rapportés dans les études cliniques ont été des affections hématologiques et du système lymphatique incluant anémie (45,7 %), neutropénie (45,3 %) et thrombopénie (27 %), des troubles généraux et anomalies au site d'administration incluant fatigue (28,3 %), fièvre (21 %) et œdème périphérique (13 %) et des infections et infestations incluant pneumonie (10,7 %). Des effets indésirables de neuropathie périphérique ont été rapportés chez 12,3 % des patients et des événements thromboemboliques veineux (ETV) chez 3,3 % des patients. Les effets indésirables de grade 3 ou 4 les plus fréquemment rapportés ont été des affections hématologiques et du système lymphatique incluant neutropénie (41,7 %), anémie (27 %) et thrombopénie (20,7 %), des infections et infestations incluant pneumonie (9 %) et des troubles généraux et anomalies au site d'administration incluant fatigue (4,7 %), fièvre (3 %) et œdème périphérique (1,3 %). L'effet indésirable grave le plus fréquemment rapporté a été la pneumonie (9,3 %). Les autres effets indésirables graves rapportés ont été la neutropénie fébrile (4,0 %), la neutropénie (2,0 %), la thrombopénie (1,7 %) et les ETV (1,7 %).
Les effets indésirables ont eu tendance à survenir plus fréquemment au cours des 2 premiers cycles de traitement par pomalidomide.
Tableau listant les effets indésirables
Les effets indésirables observés chez les patients traités par le pomalidomide en association avec le bortézomib et la dexaméthasone, le pomalidomide en association avec la dexaméthasone et les effets indésirables notifiés dans le cadre de la pharmacovigilance sont présentés dans le tableau 7 ci-dessous par classe de systèmes d'organes (SOC) et par fréquence pour l'ensemble des effets indésirables et des effets indésirables de grade 3 ou 4.
Les fréquences sont définies conformément aux recommandations en vigueur comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 7. Effets indésirables (EI) rapportés au cours des études cliniques et depuis la mise sur le marché
|
Traitement en association |
Pomalidomide/ bortézomib/dexaméthasone |
Pomalidomide/ dexaméthasone |
||
|
Classe de systèmes d'organes/Terme préférentiel |
EI de tous grades |
EI de grades 3−4 |
EI de tous grades |
EI de grades 3−4 |
|
Infections et infestations |
||||
|
Pneumonie |
Très fréquent |
Très fréquent |
- |
- |
|
Pneumonie (infections bactériennes, virales et fongiques, y compris infections opportunistes) |
- |
- |
Très fréquent |
Fréquent |
|
Bronchite |
Très fréquent |
Fréquent |
Fréquent |
Peu fréquent |
|
Infection des voies aériennes supérieures |
Très fréquent |
Fréquent |
Fréquent |
Fréquent |
|
Infection virale des voies aériennes supérieures |
Très fréquent |
- |
- |
- |
|
Sepsis |
Fréquent |
Fréquent |
- |
- |
|
Choc septique |
Fréquent |
Fréquent |
- |
- |
|
Sepsis neutropénique |
- |
- |
Fréquent |
Fréquent |
|
Colite à Clostridium difficile |
Fréquent |
Fréquent |
- |
- |
|
Bronchopneumonie |
- |
- |
Fréquent |
Fréquent |
|
Infection de l'appareil respiratoire |
Fréquent |
Fréquent |
Fréquent |
Fréquent |
|
Infections des voies aériennes inférieures |
Fréquent |
Fréquent |
- |
- |
|
Infection pulmonaire |
Fréquent |
Peu fréquent |
- |
- |
|
Grippe |
Très fréquent |
Fréquent |
- |
- |
|
Bronchiolite |
Fréquent |
Fréquent |
- |
- |
|
Infection des voies urinaires |
Très fréquent |
Fréquent |
- |
- |
|
Rhinopharyngite |
- |
- |
Fréquent |
- |
|
Zona |
- |
- |
Fréquent |
Peu fréquent |
|
Réactivation de l'hépatite B |
- |
- |
Fréquence indéterminée* |
Fréquence indéterminée* |
|
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) |
||||
|
Carcinome basocellulaire |
Fréquent |
Peu fréquent |
- |
- |
|
Epithélioma basocellulaire |
- |
- |
Peu fréquent |
Peu fréquent |
|
Carcinome spinocellulaire |
- |
- |
Peu fréquent |
Peu fréquent |
|
Affections hématologiques et du système lymphatique |
||||
|
Neutropénie |
Très fréquent |
Très fréquent |
Très fréquent |
Très fréquent |
|
Thrombopénie |
Très fréquent |
Très fréquent |
Très fréquent |
Très fréquent |
|
Leucopénie |
Très fréquent |
Fréquent |
Très fréquent |
Fréquent |
|
Anémie |
Très fréquent |
Très fréquent |
Très fréquent |
Très fréquent |
|
Neutropénie fébrile |
Fréquent |
Fréquent |
Fréquent |
Fréquent |
|
Lymphopénie |
Fréquent |
Fréquent |
- |
- |
|
Pancytopénie |
- |
- |
Fréquent* |
Fréquent* |
|
Affections du système immunitaire |
||||
|
Angiœdème |
- |
- |
Fréquent* |
Peu fréquent* |
|
Urticaire |
- |
- |
Fréquent* |
Peu fréquent* |
|
Réaction anaphylactique |
Fréquence indéterminée* |
Fréquence indéterminée* |
- |
- |
|
Rejet de greffe d'organe plein |
Fréquence indéterminée* |
- |
- |
- |
|
Affections endocriniennes |
||||
|
Hypothyroïdie |
Peu fréquent* |
- |
- |
- |
|
Troubles du métabolisme et de la nutrition |
||||
|
Hypokaliémie |
Très fréquent |
Fréquent |
- |
- |
|
Hyperglycémie |
Très fréquent |
Fréquent |
- |
- |
|
Hypomagnésémie |
Fréquent |
Fréquent |
- |
- |
|
Hypocalcémie |
Fréquent |
Fréquent |
- |
- |
|
Hypophosphatémie |
Fréquent |
Fréquent |
- |
- |
|
Hyperkaliémie |
Fréquent |
Fréquent |
Fréquent |
Fréquent |
|
Hypercalcémie |
Fréquent |
Fréquent |
- |
- |
|
Hyponatrémie |
- |
- |
Fréquent |
Fréquent |
|
Appétit diminué |
- |
- |
Très fréquent |
Peu fréquent |
|
Hyperuricémie |
- |
- |
Fréquent* |
Fréquent* |
|
Syndrome de lyse tumorale |
- |
- |
Peu fréquent* |
Peu fréquent* |
|
Affections psychiatriques |
||||
|
Insomnie |
Très fréquent |
Fréquent |
- |
- |
|
Dépression |
Fréquent |
Fréquent |
- |
- |
|
Etat confusionnel |
- |
- |
Fréquent |
Fréquent |
|
Affections du système nerveux |
||||
|
Neuropathie périphérique sensitive |
Très fréquent |
Fréquent |
Fréquent |
Peu fréquent |
|
Sensations vertigineuses |
Très fréquent |
Peu fréquent |
Fréquent |
Peu fréquent |
|
Tremblements |
Très fréquent |
Peu fréquent |
Fréquent |
Peu fréquent |
|
Syncope |
Fréquent |
Fréquent |
- |
- |
|
Neuropathie sensitivomotrice périphérique |
Fréquent |
Fréquent |
- |
- |
|
Paresthésie |
Fréquent |
- |
- |
- |
|
Dysgueusie |
Fréquent |
- |
- |
- |
|
Diminution du niveau de conscience |
- |
- |
Fréquent |
Fréquent |
|
Hémorragie intracrânienne |
- |
- |
Fréquent* |
Peu fréquent* |
|
Accident cérébrovasculaire |
- |
- |
Peu fréquent* |
Peu fréquent* |
|
Affections oculaires |
||||
|
Cataracte |
Fréquent |
Fréquent |
- |
- |
|
Affections de l'oreille et du labyrinthe |
||||
|
Vertige |
- |
- |
Fréquent |
Fréquent |
|
Affections cardiaques |
||||
|
Fibrillation auriculaire |
Très fréquent |
Fréquent |
Fréquent* |
Fréquent* |
|
Insuffisance cardiaque |
- |
- |
Fréquent* |
Fréquent* |
|
Infarctus du myocarde |
- |
- |
Fréquent* |
Peu fréquent* |
|
Affections vasculaires |
||||
|
Thrombose veineuse profonde |
Fréquent |
Peu fréquent |
Fréquent |
Peu fréquent |
|
Hypotension |
Fréquent |
Fréquent |
- |
- |
|
Hypertension |
Fréquent |
Fréquent |
- |
- |
|
Affections respiratoires, thoraciques et médiastinales |
||||
|
Dyspnée |
Très fréquent |
Fréquent |
Très fréquent |
Fréquent |
|
Toux |
Très fréquent |
- |
Très fréquent |
Peu fréquent |
|
Embolie pulmonaire |
Fréquent |
Fréquent |
Fréquent |
Peu fréquent |
|
Epistaxis |
- |
- |
Fréquent* |
Peu fréquent* |
|
Pneumopathie interstitielle diffuse |
- |
- |
Fréquent* |
Peu fréquent* |
|
Affections gastro-intestinales |
||||
|
Diarrhée |
Très fréquent |
Fréquent |
Très fréquent |
Fréquent |
|
Vomissements |
Très fréquent |
Fréquent |
Fréquent |
Fréquent |
|
Nausées |
Très fréquent |
Peu fréquent |
Très fréquent |
Peu fréquent |
|
Constipation |
Très fréquent |
Fréquent |
Très fréquent |
Fréquent |
|
Douleurs abdominales |
Très fréquent |
Fréquent |
- |
- |
|
Douleurs abdominales hautes |
Fréquent |
Peu fréquent |
- |
- |
|
Stomatite |
Fréquent |
Peu fréquent |
- |
- |
|
Bouche sèche |
Fréquent |
- |
- |
|
|
Distension abdominale |
Fréquent |
Peu fréquent |
- |
- |
|
Hémorragies gastro-intestinales |
- |
- |
Fréquent |
Peu fréquent |
|
Affections hépatobiliaires |
||||
|
Hyperbilirubinémie |
- |
- |
Peu fréquent |
Peu fréquent |
|
Hépatite |
- |
- |
Peu fréquent* |
- |
|
Affections de la peau et du tissu sous-cutané |
||||
|
Rash |
Très fréquent |
Fréquent |
Fréquent |
Fréquent |
|
Prurit |
- |
- |
Fréquent |
- |
|
Réaction médicamenteuse avec éosinophilie et symptômes systémiques (syndrome DRESS) |
- |
- |
Fréquence indéterminée* |
Fréquence indéterminée* |
|
Nécrolyse épidermique toxique |
- |
- |
Fréquence indéterminée* |
Fréquence indéterminée* |
|
Syndrome de Stevens-Johnson |
- |
- |
Fréquence indéterminée* |
Fréquence indéterminée* |
|
Affections musculosquelettiques et du tissu conjonctif |
||||
|
Faiblesse musculaire |
Très fréquent |
Fréquent |
- |
- |
|
Dorsalgies |
Très fréquent |
Fréquent |
- |
- |
|
Douleurs osseuses |
Fréquent |
Peu fréquent |
Très fréquent |
Fréquent |
|
Contractures musculaires |
Très fréquent |
Très fréquent |
Peu fréquent |
|
|
Affections du rein et des voies urinaires |
||||
|
Insuffisance rénale aiguë |
Fréquent |
Fréquent |
- |
- |
|
Insuffisance rénale chronique |
Fréquent |
Fréquent |
- |
- |
|
Rétention urinaire |
Fréquent |
Fréquent |
Fréquent |
Peu fréquent |
|
Insuffisance rénale |
- |
- |
Fréquent |
Fréquent |
|
Affections des organes de reproduction et du sein |
||||
|
Douleurs pelviennes |
- |
- |
Fréquent |
Fréquent |
|
Troubles généraux et anomalies au site d'administration |
||||
|
Fatigue |
Très fréquent |
Fréquent |
Très fréquent |
Fréquent |
|
Fièvre |
Très fréquent |
Fréquent |
Très fréquent |
Fréquent |
|
Œdème périphérique |
Très fréquent |
Fréquent |
Très fréquent |
Fréquent |
|
Douleur thoracique non cardiaque |
Fréquent |
Fréquent |
- |
- |
|
Œdème |
Fréquent |
Fréquent |
- |
- |
|
Investigations |
||||
|
Alanine aminotransférase augmentée |
Fréquent |
Fréquent |
Fréquent |
Fréquent |
|
Poids diminué |
Fréquent |
Fréquent |
- |
- |
|
Neutrophiles diminués |
- |
- |
Fréquent |
Fréquent |
|
Globules blancs diminués |
- |
- |
Fréquent |
Fréquent |
|
Numération plaquettaire diminuée |
- |
- |
Fréquent |
Fréquent |
|
Acide urique sanguin augmenté |
- |
- |
Fréquent* |
Peu fréquent* |
|
Lésions, intoxications et complications d'interventions |
||||
|
Chute |
Fréquent |
Fréquent |
- |
- |
* Rapportés dans le cadre de la pharmacovigilance.
Description de certains effets indésirables
Les fréquences indiquées dans cette rubrique sont issues des données des études cliniques menées chez des patients recevant le traitement par pomalidomide en association avec le bortézomib et la dexaméthasone (Pom + Btz + Dex) ou avec la dexaméthasone (Pom + Dex).
Tératogénicité
Le pomalidomide est structurellement proche du thalidomide. Le thalidomide est une substance active tératogène connue chez l'être humain qui provoque des anomalies congénitales sévères, pouvant engager le pronostic vital. Chez le rat et le lapin, le pomalidomide s'est avéré tératogène lors de l'administration pendant la phase d'organogenèse majeure (voir rubriques Fertilité, grossesse et allaitement et Données de sécurité préclinique). Si le pomalidomide est pris pendant la grossesse, un effet tératogène du pomalidomide est attendu chez l'être humain (voir rubrique Mises en garde spéciales et précautions d'emploi).
Neutropénie et thrombopénie
Une neutropénie est survenue chez 54,0 % des patients (Pom + Btz + Dex) au maximum (neutropénie de grade 3 ou 4 chez 47,1 % des patients [Pom + Btz + Dex]). La neutropénie a entraîné l'arrêt du traitement par pomalidomide chez 0,7 % des patients et les cas de neutropénie grave ont été peu fréquents.
Une neutropénie fébrile (NF) a été rapportée chez 3,2 % des patients (Pom + Btz + Dex) et 6,7 % des patients (Pom + Dex) et a été rapportée comme grave chez 1,8 % des patients (Pom + Btz + Dex) et 4,0 % des patients (Pom + Dex) (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Une thrombopénie est survenue chez 27,0 % des patients (Pom + Dex) et 39,9 % des patients (Pom + Btz + Dex). La thrombopénie a été de grade 3 ou 4 chez et 20,7 % des patients (Pom + Dex) et 28,1 % des patients (Pom + Btz + Dex), a entraîné l'arrêt du traitement par pomalidomide chez 0,7 % des patients (Pom + Btz + Dex) et 0,7 % des patients (Pom + Dex) et a été grave chez 0,7 % des patients (Pom + Btz + Dex) et 1,7 % des patients (Pom + Dex) (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
La neutropénie et la thrombopénie ont eu tendance à survenir plus fréquemment au cours des 2 premiers cycles de traitement par le pomalidomide en association avec le bortézomib et la dexaméthasone ou avec la dexaméthasone.
Infections
Les infections ont été l'effet indésirable non hématologique le plus fréquent.
Une infection est survenue chez 83,1 % des patients (Pom + Btz + Dex) et 55,0 % des patients (Pom + Dex) (de grade 3 ou 4 chez 34,9 % des patients [Pom + Btz + Dex] et 24,0 % des patients [Pom + Dex]). L'infection des voies aériennes supérieures et la pneumonie ont été les infections les plus fréquemment rapportées. Des infections fatales (de grade 5) sont survenues chez 2,7 % des patients (Pom + Dex) et 4,0 % des patients (Pom + Btz + Dex). Les infections ont entraîné un arrêt du traitement par pomalidomide chez 2,0 % des patients (Pom + Dex) et 3,6 % des patients (Pom + Btz + Dex).
Evénements thromboemboliques
Une prophylaxie par l'acide acétylsalicylique (et d'autres anticoagulants chez les patients à haut risque) était requise chez tous les patients dans les études cliniques. Un traitement anticoagulant (sauf si contre-indiqué) est recommandé (voir rubrique Mises en garde spéciales et précautions d'emploi).
Des événements thromboemboliques veineux (ETV) sont survenus chez 3,3 % des patients (Pom + Dex) et 12,2 % des patients (Pom + Btz + Dex) (de grade 3 ou 4 chez 1,3 % des patients [Pom + Dex] et 5,8 % des patients [Pom + Btz + Dex]). Des ETV ont été décrits comme graves chez 1,7 % des patients (Pom + Dex) et 4,7 % des patients (Pom + Btz + Dex), aucun événement fatal n'a été rapporté et les ETV ont été associés à un arrêt du traitement par pomalidomide chez 2,2 % des patients (Pom + Btz + Dex) au maximum.
Neuropathie périphérique
- Le pomalidomide en association avec le bortézomib et la dexaméthasone
Les patients présentant une neuropathie périphérique de grade ≥ 2 associée à des douleurs dans les 14 jours précédant la randomisation n'ont pas été inclus dans les essais cliniques. Une neuropathie périphérique est survenue chez 55,4 % des patients (10,8 % de grade 3 ; 0,7 % de grade 4). Les taux ajustés selon l'exposition étaient comparables entre les groupes de traitement.
Environ 30 % des patients présentant une neuropathie périphérique avaient des antécédents de neuropathie à l'inclusion. La neuropathie périphérique a entraîné l'arrêt du bortézomib chez environ 14,4 % des patients, l'arrêt du pomalidomide chez 1,8 % des patients et l'arrêt de la dexaméthasone chez 1,8 % des patients dans le bras Pom + Btz + Dex et 8,9 % des patients dans le bras Btz + Dex.
- Neuropathie périphérique — Le pomalidomide en association avec la dexaméthasone
Les patients présentant une neuropathie périphérique de grade ≥ 2 n'ont pas été inclus dans les études cliniques. Une neuropathie périphérique est survenue chez 12,3 % des patients (de grade 3 ou 4 chez 1,0 % des patients). Aucun événement de neuropathie périphérique n'a été rapporté comme grave et la neuropathie périphérique a entraîné l'arrêt du traitement chez 0,3 % des patients (voir rubrique Mises en garde spéciales et précautions d'emploi).
Hémorragies
Des troubles hémorragiques ont été rapportés avec le pomalidomide, en particulier chez des patients présentant des facteurs de risque tels que l'utilisation concomitante de médicaments augmentant le risque de saignements. Les événements hémorragiques incluaient des épistaxis, hémorragies intracrâniennes et hémorragies gastro-intestinales.
Réactions allergiques et réactions cutanées sévères
Des cas d'angiœdème, de réaction anaphylactique et des réactions cutanées sévères, telles que le SSJ, la NET et le syndrome DRESS, ont été rapportés avec l'utilisation du pomalidomide. Les patients présentant des antécédents de rash sévère associé au traitement par lénalidomide ou thalidomide ne doivent pas recevoir le pomalidomide (voir rubrique Mises en garde spéciales et précautions d'emploi).
Population pédiatrique
Les effets indésirables rapportés chez les patients pédiatriques (âgés de 4 à 18 ans) présentant des tumeurs cérébrales récurrentes ou progressives étaient cohérents avec le profil de sécurité connu du pomalidomide chez les patients adultes (voir rubrique Propriétés pharmacodynamiques).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
AVANT INSTAURATION du traitement par pomalidomide :
- test de grossesse lors de la consultation ou dans les 3 jours précédant la consultation si la patiente utilise une contraception efficace depuis au moins 4 semaines. Le test doit confirmer que la patiente n'est pas enceinte au moment où elle débute le traitement;
- hémogramme complet;
- bilan thyroïdien;
- sérologie du virus de l'hépatite B.
SURVEILLANCE du traitement :
- test de grossesse toutes les 4 semaines et jusqu'au moins 4 semaines après l'arrêt du traitement, sauf en cas de stérilisation tubaire confirmée;
- hémogramme complet une fois par semaine pendant les 8 premières semaines, puis une fois par mois;
- apparition d'effets indésirables hématologiques, de signes évocateurs d'une hémorragie;
- paramètres de la fonction hépatique pendant les six premiers mois de traitement, puis, ensuite selon les données cliniques du patient;
- développement de cancers secondaires;
- fonction thyroïdienne.
REMETTRE au patient la brochure appropriée d'information pour les patients, la carte d'information destinée aux patients et/ou un document équivalent en fonction du système national de carte-patient utilisé.
MEDICAMENT TERATOGENE :
- Chezla FEMME EN AGE DE PROCREER, le
médicament est contre-indiqué à moins que toutes les conditions suivantes soient
remplies :
. la patiente comprend les risques tératogènes attendus pour l'enfant à naître
. elle comprend la nécessité d'une contraception efficace, sans interruption, débutée au moins 4 semaines avant le traitement, poursuivie pendant toute la durée du traitement et au moins 4 semaines après la fin de celui-ci
. même en cas d'aménorrhée, toute femme en âge de procréer doit suivre tous les conseils pour une contraception efficace
. elle doit être en mesure de respecter les mesures de contraception efficaces
. elle est informée et comprend les conséquences potentielles d'une grossesse et la nécessité de consulter rapidement un médecin s'il existe un risque de grossesse
. elle comprend la nécessité de commencer le traitement dès que le pomalidomide lui a été délivré suite à un test de grossesse négatif
. elle comprend la nécessité et accepte de faire un test de grossesse au moins toutes les 4 semaines durant le traitement, sauf en cas de stérilisation tubaire confirmée
. elle confirme avoir bien compris les risques et les mesures de précaution nécessaires pour l'utilisation du pomalidomide.
- Pour les femmes en âge de procréer, S'ASSURER que :
. la patiente remplit les conditions requises par le programme de prévention de la grossesse, y compris une bonne compréhension du risque,
. la patiente confirme avoir compris les conditions susmentionnées.
- PATIENTS MASCULINS traités doivent remplir les conditions suivantes :
. ils comprennent les risques tératogènes attendus en cas de rapport sexuel avec une femme enceinte ou en âge de procréer,
. ils comprennent qu'il est nécessaire d'utiliser des préservatifs en cas de rapport sexuel avec une femme enceinte ou en âge de procréer n'utilisant pas une contraception efficace pendant toute la durée du traitement, en cas d'interruption des prises et pendant 7 jours après l'interruption et/ou l'arrêt du traitement. Cela concerne également les hommes vasectomisés qui doivent utiliser un préservatif en cas de rapport sexuel avec une femme enceinte ou en âge de procréer car le pomalidomide peut être présent dans le liquide séminal malgré l'absence de spermatozoïdes.
. ils comprennent qu'en cas de survenue d'une grossesse chez leur partenaire pendant le traitement par le pomalidomide ou pendant 7 jours après l'arrêt du traitement, ils doivent informer immédiatement leur médecin traitant qui devra recommander d'adresser leur partenaire à un médecin spécialiste ou expérimenté en tératologie pour évaluation et conseil.
Restrictions de prescription et de délivrance :
Dans l'idéal, le test de grossesse, la prescription et la délivrance du médicament doivent avoir lieu le même jour. La délivrance du pomalidomide aux femmes en âge de procréer doit avoir lieu dans les 7 jours suivant la prescription et après un test de grossesse négatif effectué sous contrôle médical. La prescription doit être limitée à une durée de traitement de 4 semaines au maximum conformément aux schémas posologiques dans les indications autorisées chez les femmes en âge de procréer et de 12 semaines au maximum chez tous les autres patients.
- test de grossesse lors de la consultation ou dans les 3 jours précédant la consultation si la patiente utilise une contraception efficace depuis au moins 4 semaines. Le test doit confirmer que la patiente n'est pas enceinte au moment où elle débute le traitement;
- hémogramme complet;
- bilan thyroïdien;
- sérologie du virus de l'hépatite B.
SURVEILLANCE du traitement :
- test de grossesse toutes les 4 semaines et jusqu'au moins 4 semaines après l'arrêt du traitement, sauf en cas de stérilisation tubaire confirmée;
- hémogramme complet une fois par semaine pendant les 8 premières semaines, puis une fois par mois;
- apparition d'effets indésirables hématologiques, de signes évocateurs d'une hémorragie;
- paramètres de la fonction hépatique pendant les six premiers mois de traitement, puis, ensuite selon les données cliniques du patient;
- développement de cancers secondaires;
- fonction thyroïdienne.
REMETTRE au patient la brochure appropriée d'information pour les patients, la carte d'information destinée aux patients et/ou un document équivalent en fonction du système national de carte-patient utilisé.
MEDICAMENT TERATOGENE :
- Chez
. la patiente comprend les risques tératogènes attendus pour l'enfant à naître
. elle comprend la nécessité d'une contraception efficace, sans interruption, débutée au moins 4 semaines avant le traitement, poursuivie pendant toute la durée du traitement et au moins 4 semaines après la fin de celui-ci
. même en cas d'aménorrhée, toute femme en âge de procréer doit suivre tous les conseils pour une contraception efficace
. elle doit être en mesure de respecter les mesures de contraception efficaces
. elle est informée et comprend les conséquences potentielles d'une grossesse et la nécessité de consulter rapidement un médecin s'il existe un risque de grossesse
. elle comprend la nécessité de commencer le traitement dès que le pomalidomide lui a été délivré suite à un test de grossesse négatif
. elle comprend la nécessité et accepte de faire un test de grossesse au moins toutes les 4 semaines durant le traitement, sauf en cas de stérilisation tubaire confirmée
. elle confirme avoir bien compris les risques et les mesures de précaution nécessaires pour l'utilisation du pomalidomide.
- Pour les femmes en âge de procréer, S'ASSURER que :
. la patiente remplit les conditions requises par le programme de prévention de la grossesse, y compris une bonne compréhension du risque,
. la patiente confirme avoir compris les conditions susmentionnées.
- PATIENTS MASCULINS traités doivent remplir les conditions suivantes :
. ils comprennent les risques tératogènes attendus en cas de rapport sexuel avec une femme enceinte ou en âge de procréer,
. ils comprennent qu'il est nécessaire d'utiliser des préservatifs en cas de rapport sexuel avec une femme enceinte ou en âge de procréer n'utilisant pas une contraception efficace pendant toute la durée du traitement, en cas d'interruption des prises et pendant 7 jours après l'interruption et/ou l'arrêt du traitement. Cela concerne également les hommes vasectomisés qui doivent utiliser un préservatif en cas de rapport sexuel avec une femme enceinte ou en âge de procréer car le pomalidomide peut être présent dans le liquide séminal malgré l'absence de spermatozoïdes.
. ils comprennent qu'en cas de survenue d'une grossesse chez leur partenaire pendant le traitement par le pomalidomide ou pendant 7 jours après l'arrêt du traitement, ils doivent informer immédiatement leur médecin traitant qui devra recommander d'adresser leur partenaire à un médecin spécialiste ou expérimenté en tératologie pour évaluation et conseil.
Restrictions de prescription et de délivrance :
Dans l'idéal, le test de grossesse, la prescription et la délivrance du médicament doivent avoir lieu le même jour. La délivrance du pomalidomide aux femmes en âge de procréer doit avoir lieu dans les 7 jours suivant la prescription et après un test de grossesse négatif effectué sous contrôle médical. La prescription doit être limitée à une durée de traitement de 4 semaines au maximum conformément aux schémas posologiques dans les indications autorisées chez les femmes en âge de procréer et de 12 semaines au maximum chez tous les autres patients.
ARRETER LE TRAITEMENT ET CONSULTER IMMEDIATEMENT LE MEDECIN en cas de :
- fièvre, frissons, maux de gorge, toux, aphtes ou tout autre signe d'infection ;
- saignements ou bleus inexpliqués, y compris saignements de nez et saignements des intestins ou de l'estomac ;
- respiration rapide, pouls rapide, fièvre et frissons, peu ou pas
d'urines émises, nausées et vomissements, confusion, perte de
connaissance ;
- diarrhée sévère, persistante ou sanglante (éventuellement avec des douleurs abdominales ou de la fièvre) ;
- douleur dans la poitrine ou douleur et gonflement dans les jambes, en particulier dans le bas de la jambe ou le mollet ;
- essoufflement ;
- gonflement du visage, des lèvres, de la langue et de la gorge pouvant entraîner des difficultés pour respirer ;
- modifications de l'aspect de la peau ou apparition d'excroissances sur la peau ;
- jaunissement de la peau et du blanc de l'oeil, urines foncées,
douleur du côté droit de l'abdomen, fièvre et nausées ou vomissements ;
- éruption cutanée étendue, fièvre élevée, augmentation du volume des ganglions lymphatiques et atteinte d'autres organes.
INFORMER IMMEDIATEMENT LE MEDECIN en cas de vision trouble ou double, ou
perte de la vision, difficulté à parler, faiblesse dans un bras ou une
jambe, changement dans la façon de marcher ou problèmes d'équilibre,
engourdissement persistant, diminution ou perte de sensation, perte de
mémoire ou confusion.
NE PAS FAIRE DE DON de sang ou de sperme pendant le traitement et pendant 7 jours après la fin du traitement.
PRUDENCE EN CAS DE CONDUITE de véhicules ou utilisation de machines
(fatigue, diminution du niveau de conscience, confusion et sensations vertigineuses).
FEMME EN AGE DE PROCREER : utiliser une méthode de contraception
efficace pendant au moins 4 semaines avant le début du traitement,
pendant toute la durée de celui-ci et au moins 4 semaines après l'arrêt
du traitement par le pomalidomide, même en cas d'interruption du
traitement.
PATIENT DE SEXE MASCULIN TRAITE par pomalidomide (y compris homme
vasectomisé) : utiliser des préservatifs en cas de rapport sexuel avec
une femme enceinte ou en âge de procréer n'utilisant pas une
contraception efficace pendant toute la durée du traitement, en cas
d'interruption des prises et pendant 7 jours après l'interruption et/ou
l'arrêt du traitement.
Femmes en capacité de procréer/Contraception chez les hommes et les femmes
Les femmes en capacité de procréer doivent utiliser une méthode de contraception efficace. En cas de survenue d'une grossesse chez une femme traitée par pomalidomide, le traitement doit être arrêté et la patiente doit être adressée à un médecin spécialiste ou expérimenté en tératologie pour évaluation et conseil. En cas de survenue d'une grossesse chez la partenaire d'un homme traité par pomalidomide, il est recommandé d'adresser celle-ci à un médecin spécialiste ou expérimenté en tératologie pour évaluation et conseil. Le pomalidomide est présent dans le sperme humain. A titre de précaution, tous les hommes recevant le pomalidomide doivent utiliser des préservatifs pendant toute la durée du traitement, y compris pendant les interruptions de traitement, et pendant 7 jours après l'arrêt du traitement si leur partenaire est enceinte ou en capacité de procréer et n'utilise pas de méthode contraceptive (voir rubriques Contre-indications et Mises en garde spéciales et précautions d'emploi).
Grossesse
Un effet tératogène du pomalidomide est attendu chez l'être humain. Le pomalidomide est contre-indiqué pendant la grossesse et chez les femmes en capacité de procréer, sauf si toutes les conditions de prévention de la grossesse sont réunies (voir rubrique Contre-indications et rubrique Mises en garde spéciales et précautions d'emploi).
Allaitement
On ne sait pas si le pomalidomide est excrété dans le lait maternel. Après administration chez des rates allaitantes, le pomalidomide a été détecté dans le lait. Compte tenu du risque d'effets indésirables du pomalidomide chez le nourrisson allaité, une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre le traitement en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Le pomalidomide a eu des effets délétères sur la fertilité et s'est révélé tératogène chez l'animal. Après administration chez des lapines gestantes, le pomalidomide a traversé la barrière placentaire et a été détecté dans le sang des fœtus (voir rubrique Données de sécurité préclinique).
Effet du pomalidomide sur d'autres médicaments
Il n'est pas attendu que le pomalidomide provoque des interactions pharmacocinétiques cliniquement pertinentes en raison d'une inhibition ou induction des isoenzymes du cytochrome P450 ou d'une inhibition des transporteurs lorsqu'il est co-administré avec des substrats de ces enzymes ou transporteurs. Le potentiel de telles interactions, dont l'effet possible du pomalidomide sur la pharmacocinétique des contraceptifs oraux œstro-progestatifs, n'a pas été évalué dans le cadre d'études cliniques (voir rubrique Mises en garde spéciales et précautions d'emploi, Tératogénicité).
Effet d'autres médicaments sur le pomalidomide
Le pomalidomide est métabolisé en partie par les CYP1A2 et CYP3A4/5. C'est également un substrat de la glycoprotéine P. L'administration concomitante de pomalidomide avec le kétoconazole, un inhibiteur puissant du CYP3A4/5 et de la P-gp ou avec la carbamazépine, un inducteur puissant du CYP3A4/5, n'a pas eu d'effet cliniquement pertinent sur l'exposition au pomalidomide. L'administration concomitante de fluvoxamine, un inhibiteur puissant du CYP1A2, et de pomalidomide en présence de kétoconazole a augmenté de 107 % l'exposition moyenne au pomalidomide (intervalle de confiance à 90 % [91 % à 124 %]) par rapport au pomalidomide plus kétoconazole. Au cours d'une seconde étude menée pour évaluer la contribution d'un inhibiteur du CYP1A2 seul aux modifications du métabolisme, l'administration concomitante de la fluvoxamine seule et du pomalidomide a augmenté de 125 % l'exposition moyenne au pomalidomide (intervalle de confiance à 90 % [98 % à 157 %]) par rapport au pomalidomide administré seul. En cas d'administration concomitante d'inhibiteurs puissants du CYP1A2 (par exemple, ciprofloxacine, énoxacine et fluvoxamine) avec le pomalidomide, réduire la dose de pomalidomide de 50 %.
Dexaméthasone
L'administration concomitante de doses répétées de pomalidomide allant jusqu'à 4 mg avec 20 mg à 40 mg de dexaméthasone (un inducteur faible à modéré de plusieurs enzymes du CYP, dont le CYP3A) chez des patients atteints de myélome multiple, n'a pas eu d'effet sur la pharmacocinétique du pomalidomide par rapport à l'administration de pomalidomide seul.
Les effets de la dexaméthasone sur la warfarine ne sont pas connus. Une surveillance étroite de la concentration de la warfarine est conseillée pendant le traitement.
Le traitement doit être instauré et suivi sous la surveillance de médecins expérimentés dans la prise en charge du myélome multiple.
La posologie est ensuite maintenue ou modifiée en fonction des résultats des examens cliniques et des analyses biologiques (voir rubrique Mises en garde spéciales et précautions d'emploi).
Posologie
Le pomalidomide en association avec le bortézomib et la dexaméthasone
La dose initiale recommandée de POMALIDOMIDE REIG JOFRE est de 4 mg par voie orale une fois par jour les Jours 1 à 14 de chaque cycle de 21 jours.
Le pomalidomide est administré en association avec le bortézomib et la dexaméthasone, comme indiqué dans le tableau 1.
La dose initiale recommandée de bortézomib est de 1,3 mg/m2 par voie intraveineuse ou sous-cutanée une fois par jour, aux jours indiqués dans le tableau 1. La dose recommandée de dexaméthasone est de 20 mg, par voie orale une fois par jour, aux jours indiqués dans le tableau 1.
Le traitement par le pomalidomide en association avec le bortézomib et la dexaméthasone doit être poursuivi jusqu'à la progression de la maladie ou l'apparition d'une toxicité inacceptable.
Tableau 1. Schéma posologique recommandé pour le pomalidomide en association avec le bortézomib et la dexaméthasone
|
|
Jour (de chaque cycle de 21 jours) |
||||||||||||||||||||
|
Cycles 1-8 |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
21 |
|
Pomalidomide (4 mg) |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
|
|
|
|
|
|
|
|
Bortézomib (1,3 mg/m2) |
• |
|
|
• |
|
|
|
• |
|
|
• |
|
|
|
|
|
|
|
|
|
|
|
Dexaméthasone (20 mg)* |
• |
• |
|
• |
• |
|
|
• |
• |
|
• |
• |
|
|
|
|
|
|
|
|
|
|
|
Jour (de chaque cycle de 21 jours) |
||||||||||||||||||||
|
Cycle 9 et cycles suivants |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
21 |
|
Pomalidomide (4 mg) |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
|
|
|
|
|
|
|
|
Bortézomib (1,3 mg/m2) |
• |
|
|
|
|
|
|
• |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dexaméthasone (20 mg)* |
• |
• |
|
|
|
|
|
• |
• |
|
|
|
|
|
|
|
|
|
|
|
|
* Pour les patients dont l'âge est > 75 ans, voir « Populations particulières ».
Modification de dose ou interruption du traitement par pomalidomide
Pour commencer un nouveau cycle de pomalidomide, le taux de neutrophiles doit être ≥ 1 x 109/L et le taux de plaquettes ≥ 50 x 109/L.
Les instructions concernant les réductions de dose ou les interruptions du traitement par pomalidomide en cas d'effets indésirables sont présentées dans le tableau 2 et les paliers de dose sont définis dans le tableau 3 ci-dessous :
Tableau 2. Instructions relatives aux modifications de dose de pomalidomide∞
|
Toxicité |
Modification de la dose |
|
Neutropénie* PNN** < 0,5 x 109/L ou neutropénie fébrile (fièvre ≥ 38,5 °C et PNN < 1 x 109/L) |
Interrompre le traitement par pomalidomide pour le reste du cycle. Contrôler la NFS*** une fois par semaine. |
|
Retour des PNN ≥ 1 x 109/L |
Reprendre le traitement par pomalidomide en diminuant la dose d'un palier par rapport à la dose antérieure. |
|
Pour chaque chute ultérieure < 0,5 x 109/L |
Interrompre le traitement par pomalidomide. |
|
Retour des PNN ≥ 1 x 109/L |
Reprendre le traitement par pomalidomide en diminuant la dose d'un palier par rapport à la dose antérieure. |
|
Thrombopénie Numération plaquettaire < 25 x 109/L |
Interrompre le traitement par pomalidomide pour le reste du cycle. Contrôler la NFS*** une fois par semaine. |
|
Retour de la numération plaquettaire ≥ 50 x 109/L |
Reprendre le traitement par pomalidomide en diminuant la dose d'un palier par rapport à la dose antérieure. |
|
Pour chaque chute ultérieure < 25 x 109/L |
Interrompre le traitement par pomalidomide. |
|
Retour de la numération plaquettaire ≥ 50 x 109/L |
Reprendre le traitement par pomalidomide en diminuant la dose d'un palier par rapport à la dose antérieure. |
|
Eruption cutanée Eruption cutanée de grades 2-3 |
Envisager une interruption ou l'arrêt du traitement par pomalidomide. |
|
Eruption cutanée de grade 4 ou formation de vésicules (y compris en cas d'angiœdème, de réaction anaphylactique, de rash bulleux ou avec exfoliation ou de suspicion de syndrome de Stevens-Johnson [SSJ], de nécrolyse épidermique toxique [NET] ou d'une réaction médicamenteuse avec éosinophilie et symptômes systémiques [syndrome DRESS]) |
Arrêter définitivement le traitement (voir rubrique Mises en garde spéciales et précautions d'emploi). |
|
Autre Autres événements indésirables de grade ≥ 3 liés au pomalidomide |
Interrompre le traitement par pomalidomide pour le reste du cycle. Lors du cycle suivant, reprendre le traitement en diminuant la dose d'un palier par rapport à la dose antérieure (attendre que l'événement indésirable ait disparu ou ait régressé à un grade ≤ 2 avant de reprendre le traitement). |
∞ Les instructions relatives aux modifications de dose dans ce tableau s'appliquent au pomalidomide en association avec le bortézomib et la dexaméthasone et au pomalidomide en association avec la dexaméthasone.
* En cas de neutropénie, le médecin doit envisager l'utilisation de facteurs de croissance.
** PNN — polynucléaires neutrophiles.
*** NFS — numération formule sanguine.
Tableau 3. Réduction de la dose de pomalidomide∞
|
Palier de dose |
Dose orale de pomalidomide |
|
Dose initiale |
4 mg |
|
Palier de dose -1 |
3 mg |
|
Palier de dose -2 |
2 mg |
|
Palier de dose -3 |
1 mg |
∞ La réduction de dose dans ce tableau s'applique au pomalidomide en association avec le bortézomib et la dexaméthasone et au pomalidomide en association avec la dexaméthasone.
Si les effets indésirables réapparaissent après réduction de la dose à 1 mg, le traitement doit être arrêté.
Inhibiteurs puissants du CYP1A2
En cas d'administration concomitante d'inhibiteurs puissants du CYP1A2 (par exemple, ciprofloxacine, énoxacine et fluvoxamine) avec le pomalidomide, réduire la dose de pomalidomide de 50 % (voir rubriques Interactions avec d'autres médicaments et autres formes d'interactions et Propriétés pharmacocinétiques).
Modification de dose ou interruption du traitement par bortézomib
Pour les instructions concernant les réductions de dose ou les interruptions du traitement par bortézomib en cas d'effets indésirables, les médecins doivent se référer au Résumé des Caractéristiques du Produit (RCP) du bortézomib.
Modification de dose ou interruption du traitement par dexaméthasone
Les instructions concernant les réductions de dose ou les interruptions du traitement par dexaméthasone à faible dose en cas d'effets indésirables sont présentées dans les tableaux 4 et 5 ci-dessous. Toutefois, les décisions liées à l'interruption ou la reprise du traitement sont à l'appréciation du médecin, conformément au Résumé des Caractéristiques du Produit (RCP).
Tableau 4. Instructions relatives aux modifications de dose de dexaméthasone
|
Toxicité |
Modification de la dose |
|
Dyspepsie de grades 1-2 |
Poursuivre le traitement et traiter par des antihistaminiques (H2) ou équivalents. Diminuer la dose d'un palier si les symptômes persistent. |
|
Dyspepsie de grade ≥ 3 |
Interrompre le traitement jusqu'à la résolution des symptômes. Ajouter un antihistaminique (H2) ou équivalent et reprendre le traitement en diminuant la dose d'un palier par rapport à la dose antérieure. |
|
Œdème de grade ≥ 3 |
Utiliser des diurétiques si nécessaire et diminuer la dose d'un palier. |
|
Confusion ou altération de l'humeur de grade ≥ 2 |
Interrompre le traitement jusqu'à la résolution des symptômes. Reprendre le traitement en diminuant la dose d'un palier par rapport à la dose antérieure. |
|
Faiblesse musculaire de grade ≥ 2 |
Interrompre le traitement jusqu'à ce que la faiblesse musculaire ait régressé à un grade ≤ 1. Reprendre le traitement en diminuant la dose d'un palier par rapport à la dose antérieure. |
|
Hyperglycémie de grade ≥ 3 |
Diminuer la dose d'un palier. Traiter par insuline ou hypoglycémiants oraux si nécessaire. |
|
Pancréatite aiguë |
Retirer la dexaméthasone du schéma thérapeutique. |
|
Autres événements indésirables de grade ≥ 3 liés à la dexaméthasone |
Arrêter l'administration de dexaméthasone jusqu'à la régression de l'événement indésirable à un grade ≤ 2. Reprendre le traitement en diminuant la dose d'un palier par rapport à la dose antérieure. |
Si la récupération des toxicités est prolongée au-delà de 14 jours, la dose de dexaméthasone sera reprise en diminuant la dose d'un palier par rapport à la dose antérieure.
Tableau 5. Réduction de dose de dexaméthasone
|
Palier de dose |
≤ 75 ans Dose (Cycles 1-8 : Jours 1, 2, 4, 5, 8, 9, 11, 12 d'un cycle de 21 jours Cycle ≥ 9 : Jours 1, 2, 8, 9 d'un cycle de 21 jours) |
> 75 ans Dose (Cycles 1-8 : Jours 1, 2, 4, 5, 8, 9, 11, 12 d'un cycle de 21 jours Cycle ≥ 9 : Jours 1, 2, 8, 9 d'un cycle de 21 jours) |
|
Dose initiale |
20 mg |
10 mg |
|
Palier de dose -1 |
12 mg |
6 mg |
|
Palier de dose -2 |
8 mg |
4 mg |
Le traitement par dexaméthasone doit être interrompu si le patient ne tolère pas une dose de 8 mg s'il est âgé de ≤ 75 ans ou s'il ne tolère pas une dose de 4 mg s'il est âgé de > 75 ans.
En cas d'arrêt définitif d'un composant du schéma thérapeutique, la poursuite du traitement par les médicaments restants est à l'appréciation du médecin.
Le pomalidomide en association avec la dexaméthasone
La dose initiale recommandée de POMALIDOMIDE REIG JOFRE est de 4 mg, prise par voie orale une fois par jour les Jours 1 à 21 de chaque cycle de 28 jours.
La dose recommandée de dexaméthasone est de 40 mg prise par voie orale une fois par jour les Jours 1, 8, 15 et 22 de chaque cycle de 28 jours.
Le traitement par le pomalidomide en association avec la dexaméthasone doit être poursuivi jusqu'à la progression de la maladie ou l'apparition d'une toxicité inacceptable.
Modification de dose ou interruption du traitement par pomalidomide
Les instructions concernant les réductions de dose ou les interruptions du traitement par pomalidomide en cas d'effets indésirables sont présentées dans les tableaux 2 et 3.
Modification de dose ou interruption du traitement par dexaméthasone
Les instructions concernant les réductions de dose de dexaméthasone en cas d'effets indésirables sont présentées dans le tableau 4. Les instructions concernant les réductions de dose de la dexaméthasone en cas d'effets indésirables sont présentées dans le tableau 6 ci-dessous. Toutefois, les décisions liées à l'interruption ou la reprise du traitement sont à l'appréciation du médecin, conformément au Résumé des Caractéristiques du Produit (RCP) en vigueur.
Tableau 6. Réduction de dose de dexaméthasone
|
Palier de dose |
≤ 75 ans Jours 1, 8, 15 et 22 de chaque cycle de 28 jours |
> 75 ans Jours 1, 8, 15 et 22 de chaque cycle de 28 jours |
|
Dose initiale |
40 mg |
20 mg |
|
Palier de dose -1 |
20 mg |
12 mg |
|
Palier de dose -2 |
10 mg |
8 mg |
Le traitement par la dexaméthasone doit être interrompu si le patient ne tolère pas une dose de 10 mg s'il est âgé de ≤ 75 ans ou s'il ne tolère pas une dose de 8 mg s'il est âgé de > 75 ans.
Populations particulières
Personnes âgées
Aucune adaptation de la dose de pomalidomide n'est nécessaire.
- Le pomalidomide en association avec le bortézomib et la dexaméthasone
Chez les patients dont l'âge est > 75 ans, la dose initiale de dexaméthasone est de :
- Pour les cycles 1 à 8 : 10 mg une fois par jour les Jours 1, 2, 4, 5, 8, 9, 11 et 12 de chaque cycle de 21 jours.
- Pour le cycle 9 et les cycles suivants : 10 mg une fois par jour les Jours 1, 2, 8 et 9 de chaque cycle de 21 jours.
- Le pomalidomide en association avec la dexaméthasone
Chez les patients dont l'âge est > 75 ans, la dose initiale de dexaméthasone est de :
- 20 mg une fois par jour les Jours 1, 8, 15 et 22 de chaque cycle de 28 jours.
Insuffisance hépatique
Les patients présentant un taux sérique de bilirubine totale > 1,5 x LSN (limite supérieure de la normale) n'ont pas été inclus dans les études cliniques. L'insuffisance hépatique a un effet modeste sur la pharmacocinétique du pomalidomide (voir rubrique Propriétés pharmacocinétiques). Aucune adaptation de la dose initiale de pomalidomide n'est nécessaire chez les patients présentant une insuffisance hépatique définie selon les critères de Child-Pugh. Cependant, la survenue d'effets indésirables doit être surveillée attentivement chez les patients présentant une insuffisance hépatique et une réduction de la dose ou l'interruption du traitement par pomalidomide doivent être envisagées si nécessaire.
Insuffisance rénale
Aucune adaptation de la dose de pomalidomide n'est nécessaire chez les patients présentant une insuffisance rénale. Les jours d'hémodialyse, les patients doivent prendre leur dose de pomalidomide après l'hémodialyse.
Population pédiatrique
L'utilisation du pomalidomide chez les enfants âgés de 0 à 17 ans dans l'indication du myélome multiple n'est pas justifiée.
En dehors des indications autorisées, le pomalidomide a été étudié chez des enfants âgés de 4 à 18 ans présentant des tumeurs cérébrales récurrentes ou progressives. Toutefois, les résultats des études n'ont pas permis de conclure que les bénéfices obtenus de cette utilisation dépassaient les risques encourus. Les données actuellement disponibles sont décrites aux rubriques Effets indésirables, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques.
Mode d'administration
Voie orale.
Les gélules de POMALIDOMIDE REIG JOFRE doivent être prises par voie orale, chaque jour, environ à la même heure. Les gélules ne doivent être ni ouvertes, ni cassées, ni mâchées (voir rubrique Précautions particulières d'élimination et de manipulation). Elles doivent être avalées entières, de préférence avec de l'eau, au cours ou en dehors des repas. Si le patient oublie de prendre une dose de pomalidomide pendant une journée, il doit prendre la dose normale prescrite à l'heure habituelle le lendemain. Les patients ne doivent pas ajuster la dose pour compenser une dose omise les jours précédents.
Il est recommandé d'appuyer uniquement sur une extrémité de la gélule pour l'extraire de la plaquette, ce qui réduit le risque de déformation ou de rupture de la gélule.
Durée de conservation :
4 ans
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
Dans les études, le pomalidomide a été administré à des doses uniques allant jusqu'à 50 mg chez des volontaires sains. Aucun effet indésirable grave lié à un surdosage n'a été rapporté. Au cours des études, des doses répétées allant jusqu'à 10 mg une fois par jour ont été administrées à des patients atteints d'un myélome multiple. Aucun effet indésirable grave lié à un surdosage n'a été rapporté. La myélosuppression était la toxicité dose-limitante. Dans les études, il a été observé que le pomalidomide était éliminé par hémodialyse.
En cas de surdosage, un traitement de soutien est recommandé.
Classe pharmacothérapeutique : Immunosuppresseurs, Autres immunosuppresseurs, code ATC : L04AX06.
Mécanisme d'action
Le pomalidomide a un effet cytotoxique direct contre le myélome, des effets immunomodulateurs et il inhibe le support des cellules stromales pour la prolifération des cellules malignes du myélome multiple. En particulier, le pomalidomide inhibe la prolifération et induit l'apoptose des cellules malignes hématopoïétiques. En outre, le pomalidomide inhibe la prolifération des lignées cellulaires de myélome multiple résistantes au lénalidomide et exerce un effet synergique avec la dexaméthasone dans les lignées cellulaires sensibles et résistantes au lénalidomide pour induire l'apoptose des cellules malignes. Le pomalidomide stimule l'immunité impliquant les lymphocytes T et les cellules tueuses naturelles (NK) et inhibe la synthèse de cytokines pro inflammatoires (par exemple TNF-α et IL-6) par les monocytes. Le pomalidomide inhibe également l'angiogenèse en bloquant la migration et l'adhésion des cellules endothéliales.
Le pomalidomide se lie directement à la protéine céréblon (CRBN), qui fait partie d'un complexe E3-ligase comprenant les protéines DDB1 (deoxyribonucleic acid [DNA] damage-binding protein 1), cullin-4 (CUL4) et régulateur de cullins-1 (Roc1), et pouvant inhiber l'auto-ubiquitination du CRBN dans le complexe. Les ubiquitines ligases E3 sont responsables de la polyubiquitination de différentes protéines substrats et peuvent partiellement expliquer les effets pléiotropes observés sur les cellules dans le cadre d'un traitement par pomalidomide.
En présence de pomalidomide in vitro, les protéines substrats Aiolos et Ikaros sont ciblées pour l'ubiquitination et la dégradation qui en découle, ce qui entraîne des effets cytotoxiques directs et des effets immunomodulateurs. In vivo, un traitement par pomalidomide entraîne une diminution des taux d'Ikaros chez les patients présentant un myélome multiple en rechute, réfractaires au lénalidomide.
Efficacité et sécurité cliniques
Le pomalidomide en association avec le bortézomib et la dexaméthasone
L'efficacité et la sécurité du pomalidomide en association avec le bortézomib et la dexaméthasone à faible dose (Pom + Btz + Dex-DF) ont été évaluées par rapport à celles du bortézomib en association avec la dexaméthasone à faible dose (Btz + Dex-DF) au cours d'une étude de phase III multicentrique, randomisée, en ouvert (CC-4047-MM-007) chez des patients adultes présentant un myélome multiple qui avaient reçu au moins un traitement antérieur comportant le lénalidomide et avaient montré une progression de la maladie au cours du dernier traitement ou après celui-ci. Au total, 559 patients ont été recrutés et randomisés dans l'étude : 281 dans le groupe Pom + Btz + Dex-DF et 278 dans le groupe Btz + Dex-DF. 54 % des patients étaient des hommes ; l'âge médian de la population globale était de 68 ans (min, max : 27, 89 ans). Environ 70 % des patients étaient réfractaires au lénalidomide (71,2 % dans le groupe Pom + Btz + Dex-DF, 68,7 % dans le groupe Btz + Dex-DF). Environ 40 % des patients étaient en 1re rechute et environ 73 % des patients avaient reçu un traitement antérieur à base de bortézomib.
Les patients du groupe Pom + Btz + Dex-DF ont reçu 4 mg de pomalidomide par voie orale les Jours 1 à 14 de chaque cycle de 21 jours. Le bortézomib (1,3 mg/m2/dose) a été administré aux patients dans les deux bras de l'étude les Jours 1, 4, 8 et 11 des 21 jours des cycles 1 à 8, ainsi que les Jours 1 et 8 des 21 jours du cycle 9 et des cycles suivants. La dexaméthasone à faible dose (20 mg/jour [≤ 75 ans] ou 10 mg/jour [> 75 ans]) a été administrée aux patients des deux bras de l'étude les Jours 1, 2, 4, 5, 8, 9, 11 et 12 d'un cycle de 21 jours pour les cycles 1 à 8 et les Jours 1, 2, 8 et 9 des 21 jours du cycle 9 et des cycles suivants. En cas de besoin, les doses ont été réduites et le traitement a été temporairement interrompu ou arrêté pour gérer la toxicité (voir rubrique Posologie et mode d'administration).
Le critère principal d'évaluation de l'efficacité était la survie sans progression (SSP), déterminée par l'analyse du Comité d'adjudication indépendant (IRAC) selon les critères IMWG en utilisant la population en intention de traiter (ITT). Dans le bras Pom + Btz + Dex-DF, après une durée médiane de suivi de 15,9 mois, la durée médiane de la SSP a été de 11,20 mois (IC à 95 % : 9,66 ; 13,73). Dans le bras Btz + Dex-DF, la durée médiane de la SSP a été de 7,1 mois (IC à 95 % : 5,88 ; 8,48).
Une synthèse des données d'efficacité globales est présentée dans le tableau 8, à la date du gel des données, le 26 octobre 2017. La figure 1 présente la courbe de Kaplan-Meier pour la SSP dans la population ITT.
Tableau 8. Synthèse des données d'efficacité globales
|
|
Pom + Btz + Dex-DF (N = 281) |
Btz + Dex-DF (N = 278) |
|
SSP (mois) |
|
|
|
Durée médianea (IC à 95 %)b |
11,20 (9,66 ; 13,73) |
7,10 (5,88 ; 8,48) |
|
RRc (IC à 95 %), valeur de pd |
0,61 (0,49 ; 0,77), < 0,0001 |
|
|
TRG, n (%) |
82,2 % |
50,0 % |
|
RCs |
9 (3,2) |
2 (0,7) |
|
RC |
35 (12,5) |
9 (3,2) |
|
TBRP |
104 (37,0) |
40 (14,4) |
|
RP |
83 (29,5) |
88 (31,7) |
|
RC (IC à 95 %)e, valeur de pf |
5,02 (3,35 ; 7,52), < 0,001 |
|
|
DdR (mois) |
|
|
|
Durée médianea (IC à 95 %)b |
13,7 (10,94 ; 18,10) |
10,94 (8,11 ; 14,78) |
|
RRc (IC à 95 %) |
0,76 (0,56 ; 1,02) |
|
Btz = bortézomib ; IC = intervalle de confiance ; RC = réponse complète ; DdR = durée de réponse ; RR = rapport de risque ; Dex-DF = dexaméthasone à faible dose ; RC = rapport des cotes (Odds Ratio) ; TRG = taux de réponse globale ; SSP = survie sans progression ; POM = pomalidomide ; RP = réponse partielle ; RCs = réponse complète stricte ; TBRP = très bonne réponse partielle.
a La médiane est basée sur l'estimation de Kaplan-Meier.
b IC à 95 % pour la médiane.
c Basé sur le modèle à risques proportionnels de Cox.
d La valeur de p est basée sur un test du log-rank stratifié.
e Rapports des cotes (Odds Ratio) pour Pom + Btz + Dex-DF/Btz + Dex-DF.
f La valeur de p est basée sur un test CMH, avec stratification en fonction de l'âge (<= 75 ans vs > 75 ans), du nombre de traitements antérieurs du myélome (1 versus > 1) et du taux de bêta-2 microglobuline lors de la sélection (< 3,5 mg/L versus ≥ 3,5 mg/L, ≤ 5,5 mg/L versus > 5,5 mg/L).
La durée médiane de traitement a été de 8,8 mois (12 cycles de traitement) dans le groupe Pom + Btz + Dex-DF et de 4,9 mois (7 cycles de traitement) dans le groupe Btz + Dex-DF.
L'avantage en termes de SSP était plus marqué chez les patients ayant reçu une seule ligne de traitement antérieure. Chez les patients ayant reçu une ligne de traitement antérieure du myélome, la durée médiane de la SSP a été de 20,73 mois (IC à 95 % : 15,11 ; 27,99) dans le groupe Pom + Btz + Dex-DF et de 11,63 mois (IC à 95 % : 7,52 ; 15,74) dans le groupe Btz + Dex-DF. Une réduction de 46 % du risque a été observée avec le traitement par Pom + Btz + Dex-DF (RR = 0,54, IC à 95 % : 0,36 ; 0,82).
Figure 1. Survie sans progression déterminée par l'analyse de la réponse par l'IRAC selon les critères IMWG (test du log-rank stratifié) (population ITT)
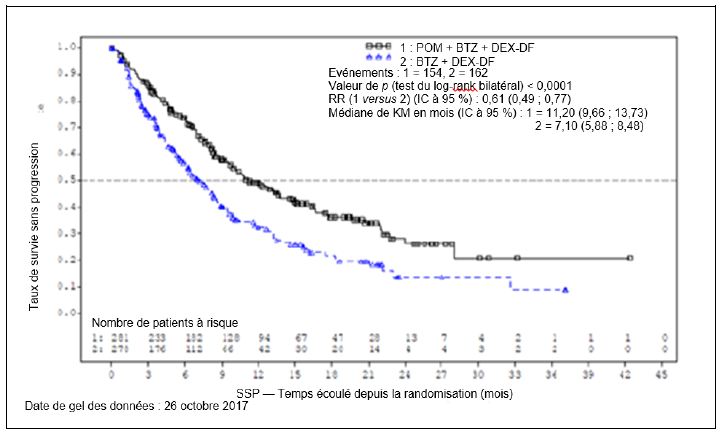
L'analyse finale de la survie globale (SG) portant sur les données disponibles au 13 mai 2022 (médiane de suivi de 64,5 mois) indique une SG médiane estimée selon la méthode de Kaplan-Meier de 35,6 mois pour le groupe Pom + Btz + Dex-DF et de 31,6 mois pour le groupe Btz + Dex-DF ; RR = 0,94, IC à 95 % : -0,77 ; 1,15, avec un taux global d'événement de 70,0 %. Aucun ajustement de l'analyse de la SG n'a été réalisé pour prendre en compte les traitements reçus ultérieurement.
Le pomalidomide en association avec la dexaméthasone
L'efficacité et la sécurité du pomalidomide en association avec la dexaméthasone ont été évaluées dans une étude de phase III multicentrique, randomisée, en ouvert (CC-4047-MM 003) au cours de laquelle le traitement par pomalidomide plus dexaméthasone à faible dose (Pom + Dex-DF) a été comparé à la dexaméthasone à dose élevée en monothérapie (Dex-DE) chez des patients adultes présentant un myélome multiple en rechute et réfractaire qui avaient reçu au moins deux traitements antérieurs comportant le lénalidomide et le bortézomib et dont la maladie avait progressé au cours du dernier traitement. Au total, 455 patients ont été recrutés dans l'étude : 302 dans le groupe Pom + Dex-DF et 153 dans le groupe Dex-DE. La majorité des patients étaient des hommes (59 %) blancs (79 %) ; l'âge médian de la population globale était de 64 ans (min, max : 35, 87 ans).
Les patients du groupe Pom + Dex-DF ont reçu 4 mg de pomalidomide par voie orale les Jours 1 à 21 de chaque cycle de 28 jours. La Dex-DF (40 mg) a été administrée une fois par jour les Jours 1, 8, 15 et 22 d'un cycle de 28 jours. Pour le bras Dex-DE, la dexaméthasone (40 mg) a été administrée une fois par jour les Jours 1 à 4, 9 à 12 et 17 à 20 d'un cycle de 28 jours. Chez les patients âgés de > 75 ans, la dose initiale de dexaméthasone était de 20 mg. Le traitement a été poursuivi jusqu'à la progression de la maladie.
Le critère principal d'évaluation de l'efficacité était la survie sans progression selon les critères de l'International Myeloma Working Group (IMWG). Pour la population en intention de traiter (ITT), la médiane de SSP, déterminée par l'analyse du Comité d'adjudication indépendant (IRAC) selon les critères IMWG a été de 15,7 semaines (IC à 95 % : 13,0 ; 20,1) dans le groupe Pom + Dex-DF ; le taux estimé de survie sans événement à 26 semaines a été de 35,99 % (± 3,46 %). Dans le bras Dex-DE, la médiane de SSP a été de 8,0 semaines (IC à 95 % : 7,0 ; 9,0) ; le taux estimé de survie sans événement à 26 semaines a été de 12,15 % (± 3,63 %).
La SSP a été évaluée dans plusieurs sous-groupes pertinents : sexe, groupe ethnique, score ECOG, facteurs de stratification (âge, populations de maladie, traitements du myélome antérieurs [2, > 2]), paramètres de significativité pronostique sélectionnés (taux initial de bêta-2 microglobuline, taux initial d'albumine, présence d'une insuffisance rénale à l'inclusion et risque cytogénétique), et exposition et absence de réponse aux traitements antérieurs du myélome. Quel que soit le groupe évalué, la SSP a généralement été concordante avec celle observée dans la population ITT pour les deux groupes de traitement.
Une synthèse de la SSP est présentée dans le tableau 9 pour la population ITT. La figure 2 présente la courbe de Kaplan-Meier pour la SSP dans la population ITT.
Tableau 9. Durée de survie sans progression déterminée par l'analyse de l'IRAC selon les critères IMWG (test du log-rank stratifié) (population ITT)
|
|
Pom + Dex-DF (N = 302) |
Dex-DE (N = 153) |
|
Survie sans progression (SSP), N |
302 (100,0) |
153 (100,0) |
|
Censurés, n (%) |
138 (45,7) |
50 (32,7) |
|
Progression/Décès, n (%) |
164 (54,3) |
103 (67,3) |
|
Durée de survie sans progression (semaines) |
||
|
Médianea |
15,7 |
8,0 |
|
IC à 95 % bilatéralb |
[13,0 ; 20,1] |
[7,0 ; 9,0] |
|
Rapport de risque (Pom + Dex-DF/Dex-DE) IC à 95 % bilatéralc |
0,45 [0,35 ; 0,59] |
|
|
Valeur de P (test du log-rank bilatéral)d |
< 0,001 |
|
Remarque : IC = intervalle de confiance ; IRAC = Independent Review Adjudication Committee, Comité d'adjudication indépendant ; NE = non estimable.
a La médiane est basée sur l'estimation de Kaplan-Meier.
b Intervalle de confiance à 95 % pour la durée médiane de survie sans progression.
c Sur la base d'un modèle de risques proportionnels de Cox comparant les fonctions de risque associées aux groupes de traitement, avec stratification en fonction de l'âge (≤ 75 versus ≥ 75), des populations de maladie (réfractaire au lénalidomide et au bortézomib versus non réfractaire aux deux substances actives) et du nombre de traitements antérieurs du myélome (2 versus > 2).
d La valeur de p est basée sur un test du log-rank stratifié selon les mêmes facteurs de stratification que le modèle de Cox ci-dessus.
Date de gel des données : 7 septembre 2012
Figure 2. Survie sans progression déterminée par l'analyse de la réponse par l'IRAC selon les critères IMWG (test du log-rank stratifié) (population ITT)
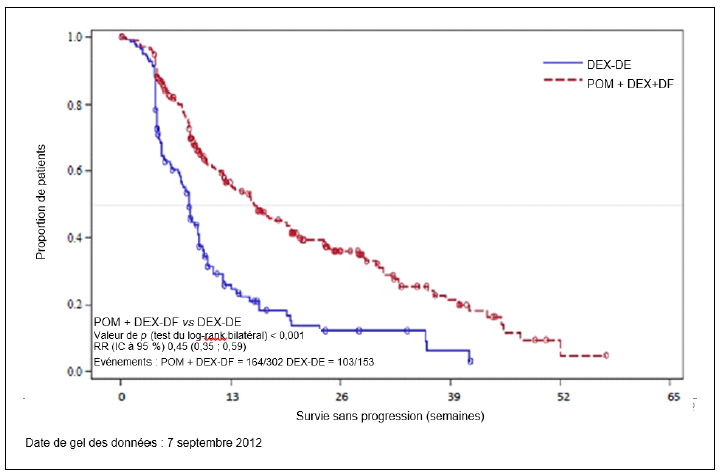
La survie globale était le principal critère d'évaluation secondaire de l'étude. Au total, 226 patients du groupe Pom + Dex-DF (74,8 %) et 95 patients du groupe Dex-DE (62,1 %) étaient en vie à la date du gel des données (7 septembre 2012). La médiane de SG des estimations de Kaplan-Meier n'a pas été atteinte pour le groupe Pom + Dex-DF, mais il est attendu qu'elle soit d'au moins 48 semaines, soit la limite inférieure de l'IC à 95 %. Dans le bras Dex-DE, la SG médiane a été de 34 semaines (IC à 95 % : 23,4 ; 39,9). Le taux sans événement à 1 an a été de 52,6 % (± 5,72 %) dans le bras Pom + Dex-DF et de 28,4 % (± 7,51 %) dans le bras Dex-DE. La différence de SG entre les deux groupes de traitement a été statistiquement significative (p < 0,001).
Une synthèse de la survie globale est présentée dans le tableau 10 pour la population ITT. La figure 3 présente la courbe de Kaplan-Meier pour la SG dans la population ITT.
Sur la base des résultats des deux critères de SSP et de SG, le Comité de surveillance indépendant établi pour cette étude a recommandé de terminer l'étude et de faire passer les patients du groupe Dex-DE dans le groupe Pom + Dex-DF.
Tableau 10. Survie globale : population ITT
|
|
Statistiques |
Pom + Dex-DF (N = 302) |
Dex-DE (N = 153) |
|
|
N |
302 (100,0) |
153 (100,0) |
|
Censurés |
n (%) |
226 (74,8) |
95 (62,1) |
|
Décédés |
n (%) |
76 (25,2) |
58 (37,9) |
|
Durée de survie (semaines) |
Médianea |
NE |
34,0 |
|
|
IC à 95 % bilatéralb |
[48,1 ; NE] |
[23,4 ; 39,9] |
|
Rapport de risque (Pom + Dex-DF/Dex-DE) [IC à 95 % bilatéralc] |
0,53 [0,37 ; 0,74] |
||
|
Valeur de P (test du log-Rank bilatéral)d |
< 0,001 |
||
Remarque : IC = intervalle de confiance. NE = non estimable.
a La médiane est basée sur l'estimation de Kaplan-Meier.
b Intervalle de confiance à 95 % pour la durée médiane de survie globale.
c Sur la base d'un modèle de risques proportionnels de Cox comparant les fonctions de risque associées aux groupes de traitement.
d La valeur de P est basée sur un test du log-rank non stratifié.
Date de gel des données : 7 septembre 2012
Figure 3. Courbe de Kaplan-Meier de la survie globale (population ITT)
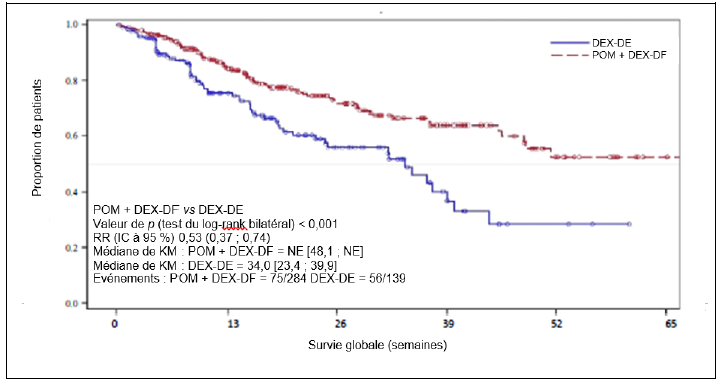
Population pédiatrique
Au cours d'une étude de phase I à dose croissante, en ouvert, à bras unique, la dose maximale tolérée (DMT) et/ou la dose recommandée pour la phase II (DRP2) du pomalidomide pour les patients pédiatriques a été déterminée à 2,6 mg/m2/jour, administrée par voie orale les Jours 1 à 21 d'un cycle de 28 jours répété.
L'efficacité n'a pas été démontrée au cours d'une étude multicentrique de phase II, en ouvert, en groupes parallèles, menée auprès de 52 patients pédiatriques âgés de 4 à 18 ans, traités par pomalidomide et présentant un gliome de haut grade, un médulloblastome, un épendymome ou un gliome infiltrant du tronc cérébral (Diffuse Intrinsic Pontine Glioma, DIPG), récurrent ou progressif, avec localisation primaire dans le système nerveux central (SNC).
Au cours de l'étude de phase II, deux patients du groupe gliome de haut grade (N = 19) ont obtenu une réponse telle que définie dans le protocole, l'un de ces patients présentant une réponse partielle (RP) et l'autre présentant une maladie stable (MS) à long terme, ayant entraîné une réponse objective (RO) et un taux de MS à long terme de 10,5 % (IC à 95 % : 1,3 ; 33,1). Un patient du groupe épendymome (N = 9) a présenté une MS à long terme, ayant entraîné une RO et un taux de MS à long terme de 11,1 % (IC à 95 % : 0,3 ; 48,2). Aucune RO confirmée ni MS à long terme n'a été observée chez les patients évaluables dans les groupes gliome infiltrant du tronc cérébral (DIPG) (N = 9) et médulloblastome (N = 9). Aucun des 4 groupes parallèles évalués dans cette étude de phase II n'a satisfait au critère d'évaluation principal de réponse objective ou de taux de maladie stable à long terme.
Le profil de sécurité globale du pomalidomide chez les patients pédiatriques était cohérent avec le profil de sécurité connu pour les adultes. Les paramètres pharmacocinétiques (PK) ont été évalués dans une analyse PK intégrée des études de phase I et de phase II et ne présentaient aucune différence significative avec ceux observés chez les patients adultes (voir rubrique Propriétés pharmacocinétiques).
Absorption
Après administration d'une dose orale unique, l'absorption du pomalidomide est d'au moins 73 % et la concentration plasmatique maximale (Cmax) est atteinte en 2 à 3 heures. L'exposition systémique (ASC : aire sous la courbe) au pomalidomide augmente de façon approximativement linéaire et dose-proportionnelle. Après administration de doses répétées, le rapport d'accumulation du pomalidomide est de 27 à 31 % sur l'ASC.
L'administration au cours d'un repas hyperlipidique et hypercalorique ralentit la vitesse d'absorption, en diminuant d'environ 27 % la Cmax moyenne, mais a un effet minimal sur l'absorption totale, avec une diminution de 8 % de l'ASC moyenne. Par conséquent, le pomalidomide peut être administré au cours ou en dehors des repas.
Distribution
Le volume de distribution apparent (Vd/F) moyen du pomalidomide est de 62 à 138 L à l'état d'équilibre. Après administration d'une dose de 2 mg une fois par jour pendant 4 jours chez des volontaires sains, le pomalidomide est distribué dans le sperme à une concentration représentant environ 67 % de la concentration plasmatique à 4 heures post-dose (approximativement le Tmax). In vitro, la liaison des énantiomères du pomalidomide aux protéines plasmatiques humaines est de 12 % à 44 % et n'est pas dépendante de la concentration.
Biotransformation
Le pomalidomide est le principal composant en circulation (environ 70 % de la radioactivité plasmatique) in vivo après administration d'une dose orale unique de [14C]-pomalidomide (2 mg) chez des volontaires sains. Aucun métabolite n'est présent à une concentration > 10 % de la radioactivité de la molécule mère ou de la radioactivité totale dans le plasma.
Les principales voies métaboliques de la radioactivité excrétée sont une hydroxylation suivie d'une glucuroconjugaison ou d'une hydrolyse. In vitro, le CYP1A2 et le CYP3A4 ont été identifiés comme les principales enzymes impliquées dans l'hydroxylation du pomalidomide induite par le CYP, avec des contributions supplémentaires minimes du CYP2C19 et du CYP2D6. Le pomalidomide est également un substrat de la glycoprotéine P in vitro. L'administration concomitante de pomalidomide avec le kétoconazole, un inhibiteur puissant du CYP3A4/5 et de la P-gp ou avec la carbamazépine, un inducteur puissant du CYP3A4/5, n'a pas eu d'effet cliniquement pertinent sur l'exposition au pomalidomide. L'administration concomitante de fluvoxamine, un inhibiteur puissant du CYP1A2, et de pomalidomide en présence de kétoconazole a augmenté de 107 % l'exposition moyenne au pomalidomide (intervalle de confiance à 90 % [91 % à 124 %]) par rapport au pomalidomide plus kétoconazole. Dans une seconde étude menée pour évaluer la contribution d'un inhibiteur du CYP1A2 seul aux modifications du métabolisme, l'administration concomitante de la fluvoxamine seule et du pomalidomide a augmenté de 125 % l'exposition moyenne au pomalidomide [intervalle de confiance à 90 %, 98 % à 157 %] par rapport au pomalidomide administré seul. En cas d'administration concomitante d'inhibiteurs puissants du CYP1A2 (par exemple, ciprofloxacine, énoxacine et fluvoxamine) avec le pomalidomide, réduire la dose de pomalidomide de 50 %. Après administration de pomalidomide chez des fumeurs, le tabagisme étant connu comme étant un inducteur de l'isoenzyme CYP1A2, il n'a pas été observé d'effet cliniquement pertinent concernant l'exposition au pomalidomide par rapport à celle observée chez les non-fumeurs.
Sur la base des données in vitro, le pomalidomide n'est pas un inhibiteur ou un inducteur des isoenzymes du cytochrome P-450 et il n'inhibe aucun des transporteurs de médicaments qui ont été étudiés. Aucune interaction cliniquement pertinente n'est attendue en cas d'administration concomitante du pomalidomide avec des substrats de ces voies.
Élimination
La demi-vie plasmatique du pomalidomide est d'environ 9,5 heures chez les volontaires sains et d'environ 7,5 heures chez les patients atteints d'un myélome multiple. La clairance corporelle totale (Cl/F) moyenne est d'environ 7 à 10 L/h.
Après administration d'une dose orale unique de [14C]-pomalidomide (2 mg) chez des volontaires sains, environ 73 % et 15 % de la dose radioactive sont éliminés dans les urines et les fèces respectivement, 2 % et 8 % du radiocarbone administré étant éliminés sous forme de pomalidomide dans les urines et les fèces.
Le pomalidomide est fortement métabolisé avant l'élimination, les métabolites en résultant étant éliminés essentiellement par voie urinaire. Les 3 métabolites majeurs présents dans les urines (formés par hydrolyse ou hydroxylation suivie d'une glucuroconjugaison) représentent environ 23 %, 17 % et 12 %, respectivement, de la dose éliminée dans les urines.
Les métabolites dépendants du CYP représentent environ 43 % de la radioactivité totale éliminée, tandis que les métabolites hydrolytiques non dépendants du CYP représentent 25 % de celle-ci, l'élimination du pomalidomide sous forme inchangée représentant 10 % (2 % dans les urines et 8 % dans les fèces).
Pharmacocinétique (PK) de population
Selon une analyse PK de population utilisant un modèle bicompartimental, la clairance apparente (Cl/F) et le volume apparent de distribution dans le compartiment central (V2/F) sont comparables chez les volontaires sains et les patients présentant un myélome multiple. Dans les tissus périphériques, le pomalidomide a été majoritairement capté par les tumeurs, avec une clairance intercompartimentale apparente (Q/F) et un volume apparent de distribution dans le compartiment périphérique (V3/F) 3,7 fois et 8 fois plus élevés, respectivement, que chez les sujets sains.
Population pédiatrique
Suite à une dose orale unique de pomalidomide administrée à des enfants et jeunes adultes présentant une tumeur cérébrale primaire récurrente ou progressive, le Tmax médian a été de 2 à 4 heures après la prise de la dose et correspondait à une moyenne géométrique des valeurs Cmax (% CV) de 74,8 (59,4 %), 79,2 (51,7 %) et 104 (18,3 %) ng/mL aux doses de 1,9, 2,6 et 3,4 mg/m2, respectivement. L'ASC0-24 et l'ASC0-inf ont suivi des tendances similaires, avec une exposition totale comprise entre 700 et 800 h•ng/mL environ aux 2 doses inférieures, et 1 200 h•ng/mL environ à la dose la plus élevée. Les estimations de la demi-vie se situaient entre 5 et 7 heures environ. Aucune tendance claire n'a pu être mise en évidence avec une stratification en fonction de l'âge ou de l'usage de stéroïdes à la DMT. Dans l'ensemble, les données suggèrent que l'ASC a augmenté de manière quasiment proportionnelle à l'augmentation de la dose de pomalidomide, alors que l'augmentation de la Cmax était généralement proportionnellement inférieure.
La pharmacocinétique du pomalidomide après des administrations orales de doses allant de 1,9 mg/m2/jour à 3,4 mg/m²/jour a été déterminée pour 70 patients âgés de 4 à 20 ans dans une analyse intégrée d'une étude de phase I et de phase II portant sur des tumeurs cérébrales pédiatriques récurrentes ou progressives. Les profils de concentrations-temps du pomalidomide ont été décrits de manière adéquate par un modèle de PK à compartiment unique avec une absorption et une élimination de premier ordre. Le pomalidomide a présenté une PK linéaire et invariante dans le temps avec une variabilité modérée. Les valeurs typiques de Cl/F, Vc/F, Ka et temps de latence du pomalidomide étaient de 3,94 L/h, 43,0 L, 1,45 h-1 et 0,454 h, respectivement. La demi-vie d'élimination terminale du pomalidomide était de 7,33 heures. Excepté pour la surface corporelle (SC), aucune des covariables testées, notamment l'âge et le sexe, n'a eu d'effet sur la PK du pomalidomide. Bien que la SC ait été identifiée comme une covariable statistiquement significative des valeurs de Cl/F et Vc/F du pomalidomide, l'impact de la SC sur les paramètres d'exposition n'a pas été considéré comme cliniquement significatif.
De manière générale, il n'existe aucune différence significative de PK du pomalidomide entre les enfants et les patients adultes.
Personnes âgées
Selon les analyses pharmacocinétiques de population chez des sujets sains et des patients présentant un myélome multiple, aucun effet significatif de l'âge (19-83 ans) sur la clairance du pomalidomide administré par voie orale n'est observé. Au cours des études cliniques, aucune adaptation posologique n'a été nécessaire chez les patients âgés (> 65 ans) exposés au pomalidomide (voir rubrique Posologie et mode d'administration).
Insuffisance rénale
Les analyses pharmacocinétiques de population ont révélé que les paramètres pharmacocinétiques du pomalidomide n'étaient pas significativement modifiés chez les patients présentant une insuffisance rénale (définie par la clairance de la créatinine ou le débit de filtration glomérulaire estimé (DFGe) par rapport aux patients dont la fonction rénale était normale (ClCr ≥ 60 mL/minute). L'exposition systémique (ASC) normalisée moyenne au pomalidomide était de 98,2 % avec un intervalle de confiance à 90 % (77,4 % à 120,6 %) chez les patients présentant une insuffisance rénale modérée (DFGe ≥ 30 et ≤ 45 mL/minute/1,73 m2) par rapport aux patients dont la fonction rénale était normale. L'exposition systémique (ASC) normalisée moyenne au pomalidomide était de 100,2 % avec un intervalle de confiance à 90 % (79,7 % à 127,0 %) chez les patients présentant une insuffisance rénale sévère ne nécessitant pas de dialyse (ClCr < 30 mL/minute ou DFGe < 30 mL/minute/1,73 m2) par rapport aux patients dont la fonction rénale était normale. L'exposition systémique (ASC) normalisée moyenne au pomalidomide était augmentée de 35,8 % avec un IC à 90 % (7,5 % à 70,0 %) chez les patients présentant une insuffisance rénale sévère nécessitant des dialyses (ClCr < 30 mL/minute nécessitant des dialyses) par rapport aux patients dont la fonction rénale était normale. Les variations moyennes de l'exposition au pomalidomide dans chacun de ces groupes d'insuffisants rénaux ne nécessitent pas d'adaptation de la dose.
Insuffisance hépatique
Les paramètres pharmacocinétiques étaient légèrement modifiés chez les patients présentant une insuffisance hépatique (définie selon les critères de Child-Pugh) par rapport aux sujets sains. Par rapport aux sujets sains, l'exposition moyenne au pomalidomide était augmentée de 51 % (intervalle de confiance à 90 % [9 % à 110 %]) chez les patients présentant une insuffisance hépatique légère, de 58 % (intervalle de confiance à 90 % [13 % à 119 %]) chez ceux présentant une insuffisance hépatique modérée et de 72 % (intervalle de confiance à 90 %, [24 % à 138 %]) chez les patients présentant une insuffisance hépatique sévère. Ces augmentations moyennes de l'exposition au pomalidomide dans chacun de ces groupes d'insuffisance hépatique ne nécessitent pas d'adaptation de la dose ou du schéma posologique (voir rubrique Posologie et mode d'administration).
Le pomalidomide a une influence mineure ou modérée sur l'aptitude à conduire des véhicules et à utiliser des machines. Des cas de fatigue, de diminution du niveau de conscience, de confusion et de sensations vertigineuses ont été rapportés lors de l'utilisation du pomalidomide. Les patients doivent être informés que s'ils présentent ces effets, ils ne doivent pas conduire de véhicules, utiliser des machines ou effectuer des activités dangereuses pendant le traitement par pomalidomide.
Etudes de toxicologie en administration répétée
Chez le rat, l'administration chronique de pomalidomide aux doses de 50, 250 et 1 000 mg/kg/jour pendant 6 mois a été bien tolérée. Aucun effet indésirable n'a été observé aux doses allant jusqu'à 1 000 mg/kg/jour (rapport d'exposition égal à 175 comparativement à une dose clinique de 4 mg).
Chez le singe, le pomalidomide a été évalué au cours d'études en administration répétée d'une durée allant jusqu'à 9 mois. Dans ces études, les singes ont présenté une sensibilité plus élevée que les rats aux effets du pomalidomide. Les principales toxicités observées chez le singe étaient associées aux systèmes hématopoïétique et lymphoréticulaire. Dans l'étude de 9 mois chez le singe aux doses de 0,05 ; 0,1 et 1 mg/kg/jour, une morbidité et l'euthanasie précoce de 6 animaux ont été observées à la dose de 1 mg/kg ; elles ont été imputées aux effets immunosuppresseurs (infection staphylococcique, diminution des lymphocytes du sang périphérique, inflammation chronique du côlon, déplétion lymphoïde à l'histologie et hypocellularité médullaire) aux expositions élevées au pomalidomide (rapport d'exposition égal à 15 comparativement à une dose clinique de 4 mg). Ces effets immunosuppresseurs ont entraîné l'euthanasie précoce de 4 singes en raison d'un mauvais état de santé (selles liquides, perte d'appétit, diminution de l'apport alimentaire et perte de poids) ; l'examen histopathologique de ces animaux a révélé une inflammation chronique du côlon et une atrophie des villosités de l'intestin grêle. Une infection staphylococcique a été observée chez 4 singes ; 3 de ces animaux ont répondu à une antibiothérapie et 1 est décédé sans traitement. De plus, des observations compatibles avec une leucémie aiguë myéloblastique ont entraîné l'euthanasie de 1 singe ; les observations cliniques et la pathologie clinique et/ou les anomalies médullaires relevées chez cet animal étaient compatibles avec une immunosuppression.
Une prolifération minime ou légère des canaux biliaires accompagnée d'augmentations des ALAT et des Gamma GT a également été observée à la dose de 1 mg/kg/jour. L'examen des animaux soumis à une période de réversibilité a indiqué que tous les effets liés au traitement étaient réversibles dans les 8 semaines suivant l'arrêt du traitement, à l'exception de la prolifération des canaux biliaires intrahépatiques observée chez 1 animal du groupe 1 mg/kg/jour. La dose sans effet nocif observé (DSENO) a été de 0,1 mg/kg/jour (rapport d'exposition égal à 0,5 comparativement à une dose clinique de 4 mg).
Génotoxicité/Carcinogénicité
Le pomalidomide n'a pas été mutagène au cours des essais de mutation sur cellules bactériennes et cellules de mammifères et n'a pas induit d'aberrations chromosomiques dans des lymphocytes de sang périphérique humains ni de formation de micronoyaux dans les érythrocytes polychromatiques dans la moelle osseuse de rats ayant reçu des doses allant jusqu'à 2 000 mg/kg/jour. Aucune étude de cancérogenèse n'a été réalisée.
Fertilité et développement embryonnaire précoce
Au cours d'une étude de fertilité et de développement embryonnaire précoce chez le rat, le pomalidomide a été administré chez les mâles et les femelles aux doses de 25, 250 et 1 000 mg/kg/jour. L'examen de l'utérus le 13e jour de gestation a révélé une diminution du nombre moyen d'embryons viables et une augmentation des échecs post-implantatoires à toutes les doses. Par conséquent, la DSENO pour ces effets était < 25 mg/kg/jour (l'ASC24h était de 39 960 ng•h/mL [nanogrammes•heure/millilitre] à cette dose la plus faible testée et le rapport d'exposition était de 99 comparativement à une dose clinique de 4 mg). Lorsque les mâles traités dans cette étude ont été accouplés avec les femelles non traitées, tous les paramètres utérins ont été comparables à ceux des témoins. Sur la base de ces résultats, les effets observés ont été imputés au traitement des femelles.
Développement embryonnaire et fœtal
Chez le rat et le lapin, le pomalidomide a été tératogène lorsqu'il a été administré pendant la phase d'organogenèse majeure. Au cours d'une étude de toxicité sur le développement embryo-fœtal chez le rat, des malformations consistant en une absence de vessie, absence de thyroïde et fusion et défaut d'alignement des vertèbres thoraciques et lombaires (arcs centraux et/ou neuraux) ont été observées à toutes les doses (25, 250 et 1 000 mg/kg/jour).
Aucune toxicité maternelle n'a été observée au cours de cette étude. Par conséquent, la DSENO maternelle a été de 1 000 mg/kg/jour et la DSENO en termes de toxicité sur le développement a été < 25 mg/kg/jour (l'ASC24h était de 34 340 ng•h/mL le 17e jour de gestation à cette dose la plus faible testée et le rapport d'exposition était de 85 comparativement à une dose clinique de 4 mg). Chez le lapin, le pomalidomide administré à des doses allant de 10 à 250 mg/kg a induit des malformations embryonnaires et fœtales. Une augmentation des anomalies cardiaques a été observée à toutes les doses, avec une incidence significativement plus élevée à la dose de 250 mg/kg/jour. Aux doses de 100 et 250 mg/kg/jour, de légères augmentations des échecs post-implantatoires et de légères diminutions du poids des fœtus ont été relevées. A la dose de 250 mg/kg/jour, les malformations fœtales consistaient en des anomalies des membres (flexion et/ou rotation des membres antérieurs et/ou postérieurs, doigts non attachés ou absents) et des malformations osseuses associées (absence d'ossification métacarpienne, défaut d'alignement des phalanges et métacarpes, doigt absent, absence d'ossification des phalanges et tibia court non ossifié ou courbé), dilatation modérée du ventricule latéral du cerveau, position anormale de l'artère sous-clavière droite, absence du lobe intermédiaire du poumon, implantation basse des reins, modifications de la morphologie hépatique, ossification absente ou incomplète du pelvis, augmentation du nombre moyen de côtes thoraciques surnuméraires et diminution du nombre moyen de tarses ossifiés. Une faible réduction de la prise de poids des mères, une diminution significative des triglycérides et une diminution significative des poids absolu et relatif de la rate ont été observées aux doses de 100 et 250 mg/kg/jour. La DSENO maternelle a été de 10 mg/kg/jour et la DSENO sur le développement a été < 10 mg/kg/jour (l'ASC24h était de 418 ng•h/mL le 19e jour de gestation à cette dose la plus faible testée, soit une valeur similaire à celle obtenue avec une dose clinique de 4 mg).
Les gélules ne doivent pas être ouvertes ou écrasées. Si la poudre de pomalidomide entre en contact avec la peau, la peau doit être lavée immédiatement et abondamment au savon et à l'eau. En cas de contact avec les muqueuses, rincer abondamment à l'eau.
Les professionnels de santé et les aidants doivent porter des gants à usage unique pour manipuler la plaquette ou la gélule. Les gants doivent ensuite être retirés avec précaution afin d'éviter une exposition cutanée, placés dans un sac plastique en polyéthylène à fermeture hermétique et éliminés conformément à la réglementation en vigueur. Les mains doivent ensuite être soigneusement lavées au savon et à l'eau. Les femmes enceintes ou qui pensent l'être ne doivent pas manipuler la plaquette ou la gélule (voir rubrique Mises en garde spéciales et précautions d'emploi).
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur. Les médicaments non utilisés doivent être rapportés à un pharmacien à la fin du traitement.
Liste I
Médicament soumis à prescription hospitalière.
Prescription réservée aux spécialistes en oncologie ou en hématologie, ou aux médecins compétents en cancérologie ou en maladies du sang.
Médicament nécessitant une surveillance particulière pendant le traitement :
- pour tous les patients : la prescription nécessite la signature de l'accord de soins et la remise d'un carnet patient.
- pour les femmes en âge de procréer :
- la prescription est limitée à une durée de traitement de 4 semaines au maximum;
- un test de grossesse doit être effectué au moins toutes les 4 semaines, dans les 3 jours précédant la prescription ; la date et le résultat de ce test doivent être mentionnés dans le carnet patient ;
- la délivrance doit être effectuée au plus tard 7 jours après la prescription et après avoir vérifié la date et le résultat du test de grossesse ; la date de délivrance doit être mentionnée dans le carnet patient.
Gélule
Tête opaque jaune et corps opaque jaune, taille d'enveloppe de gélule no 4 (environ 14 mm × 5 mm) comportant la mention « LP » imprimée à l'encre noire sur la tête et la mention « 664 » imprimée à l'encre noire sur le corps, et contenant une poudre granuleuse jaune.
Les gélules sont conditionnées sous plaquettes en (PVC/PCTFE(Aclar)/Aluminium).
Présentation :
Plaquettes (PVC/PCTFE (Aclar)/Aluminium) :
21 gélules (plaquettes)
Pomalidomide.......................................................................................................................... 1 mg
Pour une gélule.
Excipient à effet notoire :
Chaque gélule de POMALIDOMIDE REIG JOFRE 1 mg contient 25,1 mg d'isomalt.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Contenu de la gélule
Isomalt (E953), amidon de maïs prégélatinisé, fumarate de stéaryle sodique.
Enveloppe de la gélule
Gélatine, dioxyde de titane (E171), oxyde de fer jaune (E172), encre d'impression noire.
Encre d'impression
Gomme laque (E904), solution concentrée d'ammoniaque, hydroxyde de potassium, oxyde de fer noir (E172).